A Revolutionary Drug to Treat and Prevent HIV Infection
By John Raul Somoza
A two-decade research effort has paid off with a treatment that can disable the deadly virus’s capsid, the protein shell that protects its genome.
A two-decade research effort has paid off with a treatment that can disable the deadly virus’s capsid, the protein shell that protects its genome.

As scientists search for new medicines, they slog through a marathon of frustration, dead ends, and moments of great excitement. Tight-knit groups of biologists and chemists often work for years to develop therapies that can prevent, control, or cure disease. Despite that effort, success is rare: The vast majority of projects never yield a compound suitable for human testing, and even those that reach clinical trials have only a 10 to 20 percent chance of becoming an approved drug.
In June of 2010, a team I was on felt the crushing weight of those statistics. We were four years into our quest to develop a novel drug for the treatment of HIV-1 (referred to simply as HIV in this article). We had tested thousands of molecules, but none had shown any promise of becoming part of a viable new therapy. Despite hoping that we still could discover a drug that would significantly improve the treatment of HIV infection, many of us on the team worried that we would never reach our goal.
Then, in 2016, after changing our strategy, we identified a compound promising enough for clinical trials. Several years later, those trials demonstrated that our drug, lenacapavir, is effective in both the treatment and prevention of HIV infection. In December of 2022, the U.S. Food and Drug Administration (FDA) approved lenacapavir as part of a new therapy for the treatment of multidrug-resistant HIV, and in June of this year, the FDA approved a once-every-six-months injection of the drug to prevent infection.
Lenacapavir represents the latest strike against HIV in a fight that started in the 1980s when the virus was first recognized as the cause of the AIDS epidemic. Although new HIV infections have dropped by 60 percent since they reached their peak in 1995, more than one million people across the world still get infected by the virus every year, and about 630,000 die annually of HIV-related causes. And with global health programs, including those targeting HIV/AIDS, facing significant funding cuts, the progress we’ve made could slow. These statistics underscore the need for new tools to treat and prevent infections.
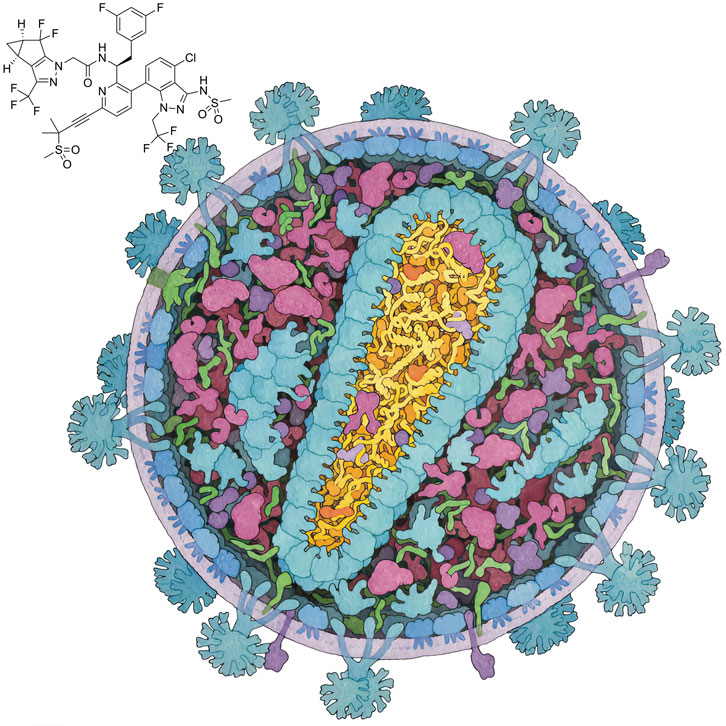
Illustration by David S. Goodsell, B-HIVE Center, RCSB Protein Data Bank, and Scripps Research
Over the decades, a number of anti-HIV drugs have been developed that disrupt the virus’s life cycle and stop it from replicating. Most of these drugs bind to and shut down viral proteins called enzymes, which catalyze biochemical reactions the virus needs to perform to replicate in a host cell. Combinations of inhibitors of these enzymes, known as combination antiretroviral therapy, are highly effective at blocking viral replication and have had an enormous impact on controlling HIV infections. Today, when used consistently, these drug combinations have transformed HIV infection from a fatal diagnosis to a manageable chronic condition. Most people on combination therapy can expect to lead normal or near-normal lives.
But a problem remains: HIV has a remarkable ability to mutate. As a viral enzyme copies HIV’s genome, it makes mistakes that result in mutations in the virus. Most of these mutations either have no effect on the virus or make it weaker. Occasionally, however, mutations arise that allow the virus to adapt to changes in its environment, enabling it to evade antiretroviral drugs. These mutations alter specific areas of the HIV proteins, making them less susceptible to binding drug molecules.
The adaptability of the virus has led to an evolutionary arms race between HIV and humans. HIV gradually becomes less susceptible to the drugs we have created, and we have had to respond by searching for drugs that attack novel proteins essential to HIV’s ability to replicate. In our team’s quest for a new HIV treatment, we decided to focus on a component of the virus that hadn’t been targeted previously: the HIV capsid, the protein shell that encloses the viral genome.
By choosing a novel target to go after, we hoped to discover an HIV treatment that got around existing resistant strains. We eventually discovered that lenacapavir successfully works as a part of such a treatment and also effectively prevents HIV infection in at-risk individuals.
We set our sights on the HIV capsid because of its essential role in the life cycle of the virus, which involves HIV infecting cells, replicating itself, and then spreading to more cells to repeat the process (see figure below). HIV is what is known as a retrovirus, meaning that it encodes its genome in RNA instead of DNA. The protein capsid encloses and protects its RNA. The capsid, in turn, is surrounded by a bubble of lipids (fats) and proteins. When HIV enters a host cell, the capsid is freed of this lipid-and-protein container, allowing it to interact with various host proteins. These protein–protein interactions help transport the capsid, along with the encased viral genome, across the cytoplasm, through a protein structure called the nuclear pore, and into the nucleus of the cell.
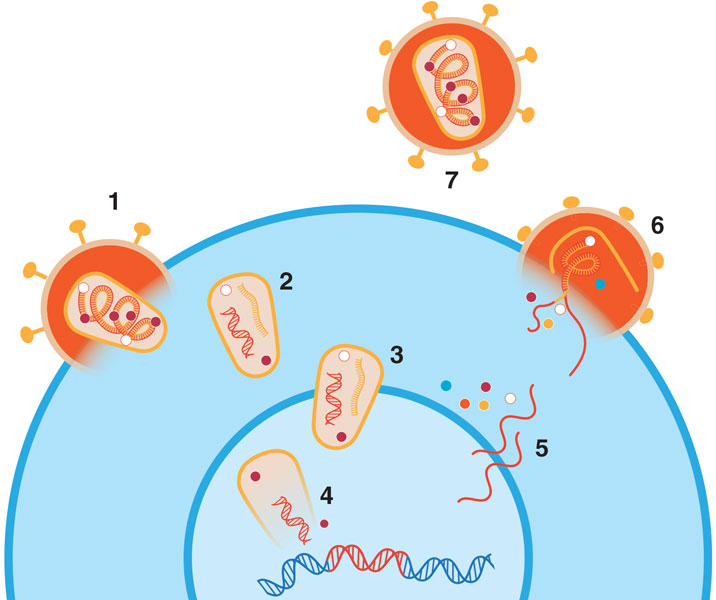
Barbara Aulicino
The virus uses an enzyme known as reverse transcriptase to convert its RNA into DNA, which then gets incorporated into the host cell genome with the help of integrase, another viral enzyme. When the infected host cell turns on its own genes, it also activates the virus’s incorporated genes, producing the RNA and proteins needed to make new viral particles.
These newly made viral components assemble near the host cell membrane and then bud out from the cell, taking part of the cell’s lipid membrane with them to help enclose the assembled viral material. At this stage, a new viral particle cannot yet infect other cells. It becomes fully mature and infectious only after protease, yet another viral enzyme, chops up the long protein chains that were created in the host cell. This enzymatic dicing transforms the chains into the virus’s final, functional proteins, including the ones that will go on to form its capsid.
The capsid is a 100-nanometer-long protein shell composed of many copies of a molecule simply called capsid protein, or CA. The CA proteins group into five- or six-member rings, creating pentamers and hexamers that interact with one another, forming a shell resembling a soccer ball that has been stretched in one direction. Some 200 to 250 hexamers and exactly 12 pentamers create the capsid, with the pentamers providing the curvature needed to close off the structure and fully enclose the viral genome.
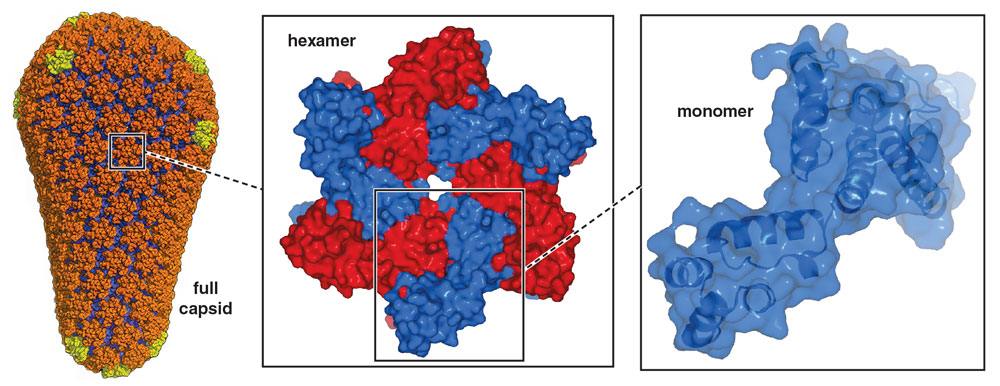
Full capsid: from Pornillos, 2011; hexamer and monomer: John Raul Somoza (hexamer and monomer structures determined by Somoza); Barbara Aulicino
For the virus to replicate itself and go on to infect other cells, it is essential that the capsid perform its functions well. The capsid must latch onto host cell proteins to be transported into the nucleus; it must undergo a well-choreographed process of disassembly to release the HIV genome; and eventually it must reassemble to form newly created viral particles.
In the spring of 2006, a group of researchers at Gilead Sciences in Foster City, California, proposed creating a new class of AIDS drugs by discovering a small molecule that could target the HIV capsid. In the world of drug discovery, small-molecule drugs are compounds that are small and “greasy” enough to penetrate cell membranes to reach targets within the cell. Based largely on the work of Wesley Sundquist’s lab at the University of Utah, the team thought that the capsid made a good target because successful replication of the virus requires the precise assembly and disassembly of this protein shell. A compound that gums up those processes—by making the shell too fragile or too strong, or by disrupting its shape—could hurt the virus’s ability to propagate.
Although new HIV infections have dropped by 60 percent since they reached their peak in 1995, more than one million people across the world still get infected by the virus every year, and about 630,000 people die annually of HIV-related causes.
The choice of capsid as a target was controversial at the time. Some scientists doubted whether it was possible to successfully identify a compound that would disrupt the capsid’s assembly and disassembly process. Up to that point, the most common drug targets for HIV had been the enzymes crucial to HIV’s life cycle: reverse transcriptase, integrase, and protease. There are several reasons why enzymes are particularly well-suited as targets for small-molecule drugs. Most significantly, enzymes are proteins that feature pockets called active sites where specific molecules bind and undergo chemical transformations. Those active sites are also suitable to bind small-molecule drugs that then block the work needed to be done by the enzyme.
But CA isn’t an enzyme. It doesn’t have pockets that have evolved to bind molecules and catalyze reactions. When CA proteins join together to form the HIV capsid shell, these proteins interact over areas much larger than that of an active site. Designing a small molecule that could bind to CA and disrupt the protein–protein interactions necessary for proper capsid function would be extremely challenging.
Despite this concern, the Gilead team decided to proceed.
Once the project was approved, I joined the team that was formed to look for compounds that impaired capsid assembly. To start this process, we developed a biochemical test, or assay, that recreated aspects of the process that occurs inside newly budded viral particles. This assay was based on the observation that in solutions with high concentrations of CA protein and sodium chloride salt, CA proteins spontaneously self-assemble to form tubelike structures that look very much like open-ended capsids. The formation of these tubes can be seen with the naked eye as the solutions become milky-white over the course of a few hours. This cloudy haze is created by the suspended tube particles in the solution. We used this tube assembly process in the lab as a surrogate for capsid formation in the virus, allowing us to identify compounds that might bind to the CA protein and disrupt capsid assembly.
Drug discovery often involves screening hundreds of thousands of molecules to find a handful that have the potential to do what we want them to do. To screen that many compounds, we developed a high-throughput assay that was both fast and capable of telling us quantitatively how effectively a test compound disrupted capsid tube formation. In the lab, we dispensed the potential inhibitor we wanted to test into transparent tubes. We then added CA protein and started the CA tube assembly by adding sodium chloride. We monitored the formation of the protein tubes quantitatively by measuring the solution’s absorbance of light at a wavelength of 350 nanometers. As the CA proteins assembled into tubes, the absorbance increased (see figure below).
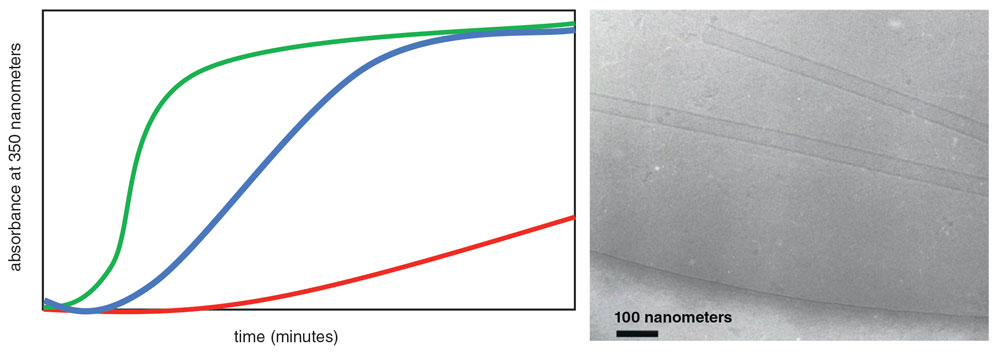
Graph: Barbara Aulicino; micrograph: Sam Li and Wes Sundquist
When testing compounds in this assay, we observed three outcomes. First, when a compound didn’t affect tube formation, we saw the same thing that happened without any compound present: Light absorbance rose steadily as the tubes assembled, eventually reaching a plateau once the majority of CA protein had assembled into tubes. These compounds weren’t what we were looking for.
Instead, we were interested in molecules that showed one of two other behaviors. For example, if adding a compound slowed or eliminated the increase in absorbance, we assumed that the compound inhibited capsid assembly. We were also interested in compounds that accelerated the rate of light absorption, although what those molecules were doing to the proteins was more difficult to interpret. We proposed that these compounds either accelerated capsid assembly or changed the morphology of the CA tubes, both of which were abilities we were interested in.
With this assay, we tested two groups of molecules. First, we synthesized and tested all compounds published in the research literature that were already implicated in binding to the HIV capsid, as well as some molecules that resembled the published ones. Also, we carried out a high-throughput screen of about 450,000 compounds from collections commonly used when screening for potential drugs.
These screens led to the discovery of a handful of compounds that affected normal capsid formation. We then wanted to see how these molecules interacted with CA in greater detail. By mixing the compounds with CA proteins and using biophysical techniques, including x-ray crystallography and nuclear magnetic resonance spectroscopy, we obtained detailed pictures of how the molecules bound to CA. From those pictures, we determined that the molecular disruptors we had identified interacted with one of two sites on the CA protein.
The first site we found, which we creatively named “Site 1,” appeared only in the presence of a molecule that to binds it. When a molecule interacts with CA, a small region of the protein rearranges to create a small pocket where the compound binds.
As we worked on characterizing and developing molecules to bind to Site 1 and disrupt capsid assembly, our initial excitement gradually gave way to frustration and doubt. There were several red flags about this site. First, we had a lot of structural information about how molecules bound to CA at Site 1 and how the area rearranged itself. But we had no clear understanding of how those local changes prevented CA proteins from assembling into larger structures.
We were also worried about how these Site 1 molecules might work against the wide range of HIV variants seen in patients. Due to HIV’s high mutation rate, different strains or variants around the globe contain proteins that vary in their sequence of amino acids, the chemical building blocks of a protein. These small molecular changes can have big consequences for drug binding. When small molecules bind to a protein, interactions with specific amino acids are often key to how the molecule latches onto the protein.
We observed that some of the amino acids that made up Site 1 were not well conserved across a broad range of HIV variants that infected patients, indicating that some people could be infected by viruses that lacked the particular amino acids necessary to bind with our candidate drug molecules. Also, we worried that this amino acid variability suggested that viruses that did bind our molecules might eventually mutate in a way that made them resistant to any potential disruptor.
Finally, and most importantly, our attempts to synthesize increasingly potent compounds targeting Site 1 eventually hit a ceiling. We could not improve the antiviral potency enough to make a useful drug. Scientists developing new medicines care about potency because a more potent molecule means a lower dose for patients. High doses lead to problems for a drug, such as large, hard-to-manage pills or a greater likelihood of causing harmful side effects. We eventually concluded that Site 1 lacked the structural features necessary to lead to a potent enough drug molecule.
In 2010, after about a year of synthesizing Site 1 compounds and about four years after the start of the project, we started to move away from Site 1 and focus our efforts on a different site on the capsid protein that we called, yes, “Site 2.” From our x-ray crystallography studies, we already had a wealth of structural information about Site 2. Individual CA proteins are grouped into hexamers that bind with neighboring hexamers. Thus, each hexamer has six identical CA–CA interfaces. One part of this interface is a deep groove that is partially formed by Site 2.
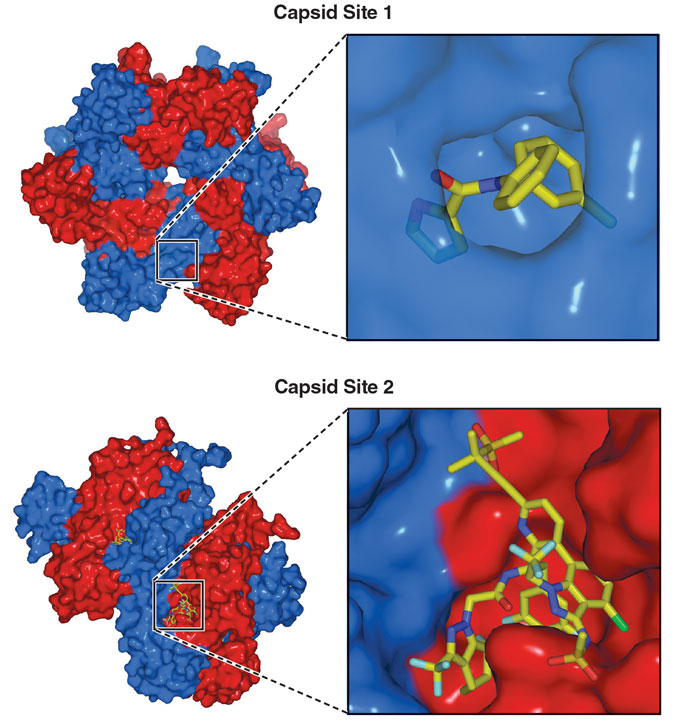
Capsid protein monomers: John Raul Somoza (structures determined by Somoza); Barbara Aulicino
In multiple ways, Site 2 seemed more promising than Site 1. Site 1 wasn’t at the interface between CA proteins, whereas Site 2 was, suggesting that a small molecule could bind to Site 2 and then either distort the CA–CA interface or stabilize it. Distorting the interface could inhibit capsid assembly, and overly stabilizing the interface could disrupt disassembly. Either alteration could throw a wrench into HIV’s life cycle.
Site 2 was also promising because there was little to no variation in the amino acids that formed it across HIV variants isolated from patients. This observation suggested that Site 2 was much less likely to undergo mutations that could make HIV resistant to our molecules. (Later, we discovered that the likely reason for this high degree of sequence conservation was that Site 2 also interacts with host cell proteins that move viral capsids through the nuclear pore and into the nucleus, a critical step in the HIV life cycle.) We also had a more instinctual reason for targeting Site 2: The deep groove that forms the site looks like the type of structural feature, like an enzyme active site, where small-molecule drugs typically bind.
An effective anti-HIV drug must satisfy two key goals. First, it needs to be sufficiently potent. Second, it must be compatible with being taken no more than once a day, like existing HIV therapies, so that it is practical for patients to take. To achieve this dosing goal, a molecule must resist metabolism in the body enough that its concentration in the bloodstream stays above a certain effective threshold throughout the day.
Although these dual requirements may seem straightforward, finding a molecule that could bind to Site 2 and that was sufficiently potent and metabolically stable took us about five years and required the synthesis, testing, and detailed characterization of thousands of candidate compounds.
On May 26, 2015, our team synthesized a compound known as GS-6207. This compound was metabolically stable and highly potent. However, it took an additional nine months of research until we could confirm that it had the potential to become a successful drug and also satisfied all the safety requirements from the FDA to be given to humans.
One remaining concern about GS-6207 was that it was less soluble than the typical drug taken as a pill, the form of most current HIV therapies. We tried to increase the solubility of GS-6207 for use in a pill, and we also tested the drug when given by injection, either intravenously or under the skin (subcutaneously). Specifically, we wanted to know how long drug levels would remain over the therapeutic threshold when delivered by injection. Would it last a day? A week? To our amazement, in animal tests, the drug remained above therapeutic levels for months when injected under the skin. In people, the drug remained safely at therapeutic levels in the bloodstream for at least 6 months.
In retrospect, we identified three factors explaining why the drug continues to work so long after injection. First, lenacapavir is extraordinarily potent. It binds very tightly to the capsid, and, as a result, a very low concentration of the drug is needed to disrupt capsid function. Second, the compound is very stable metabolically, meaning that it strongly resists the body’s attempts to degrade it. These two positive factors were the results of years of work by our team to optimize the molecule.
The third factor was the most fortuitous. When a solution or suspension of lenacapavir gets injected under the skin, the molecule comes out of solution and forms a depot of the drug. In people, this collection of drug gradually dissolves into the bloodstream over the course of months, creating a steady concentration of the compound available to inhibit capsid proteins and provide ongoing protection against HIV.
On June 30, 2016, almost 10 years after our project started, GS-6207 became known as lenacapavir and entered human clinical trials. After nearly a decade of work, our team was both excited and daunted by the prospect of our compound reaching human clinical trials. Testing a drug in humans takes years, moving through three phases of trials, first to determine the drug’s safety, and later to assess its effectiveness. Unfortunately, the majority of compounds that enter these trials are not successful, either because some unsuspected safety problem comes to light or because the drug doesn’t work as well as expected.
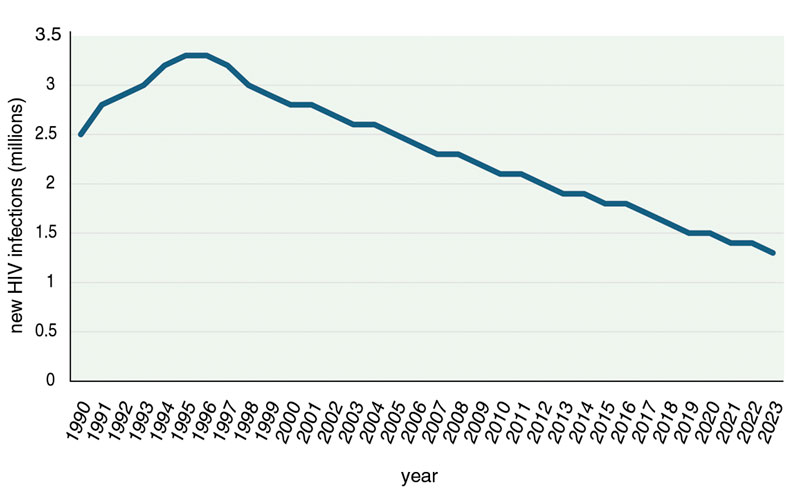
Data: UNAIDS; Barbara Aulicino
Lenacapavir’s clinical trials focused on two distinct uses of the drug: treatment and prevention of HIV infections. After successfully passing through phase 1 and 2 trials, the first phase 3 trial tested whether lenacapavir, in combination with other antiretroviral drugs, could be used to treat HIV in people who had repeatedly failed treatment due to infection with multidrug-resistant HIV variants. The use of lenacapavir to treat HIV infection had been the primary goal of our research. The results of the clinical studies showed that lenacapavir was indeed effective at treating HIV infection, even when used with drug combinations that were no longer effective on their own. By attacking a novel target, the capsid, we had developed a new treatment for multidrug-resistant HIV.
Clinical trial results in more than 4,000 people corresponded to a 96 percent reduction in risk of infection and an 89 percent improvement compared with Truvada, a commonly used once-daily pre-exposure prophylaxis pill.
The second set of phase 3 clinical trials focused on using lenacapavir to prevent HIV infection. In 2012, clinical trials had established that people who were HIV-negative but who were at risk of being exposed to the virus could prophylactically take a combination of HIV drugs to reduce the possibility of being infected. This practice is known as pre-exposure prophylaxis, and there are three drugs or drug combinations approved for this purpose: Truvada, Descovy, and Apretude.
Two phase 3 clinical trials of lenacapavir looked at the potential of twice-yearly subcutaneous injections of the drug to prevent HIV infection. In the first trial, none of the 2,134 participants receiving lenacapavir injections became infected. In the second trial, only two people out of the 2,179 participants became infected. These remarkable results in more than 4,000 people corresponded to a 96 percent reduction in risk compared with the background incidence of infection, and an 89 percent improvement compared with Truvada, a commonly used once-daily pre-exposure prophylaxis pill.
Interestingly, although lenacapavir is better at preventing infection than Truvada, we don’t think targeting the capsid is the reason it is inherently more effective. Instead, we think lenacapavir is more effective because people are better at taking the required doses of the drug than they are with Truvada. Not surprisingly, it appears to be easier to comply with a drug that gets injected every six months than with a pill that needs to be taken each and every day.
When we started this project, we were looking for a new anti-HIV drug that impaired capsid assembly, and we successfully achieved that. As we more deeply explored how lenacapavir works, we found that targeting Site 2 had some additional, unexpected benefits.
We discovered that some of the host cell proteins that help the HIV capsid cross into the nucleus of a host cell bind to the capsid through interactions with Site 2. Therefore, the effectiveness of lenacapavir is likely due to a combination of its deleterious effects on proper capsid assembly and disassembly, as well as the fact that lenacapavir binding blocks host proteins from attaching to the capsid and moving it through the nuclear pore. Although the relative importance of these two distinct mechanisms of action isn’t clear, preventing the capsid from interacting with these nuclear pore proteins is likely playing an important role in how well lenacapavir works.
This other mechanism of action also diminishes the virus’s ability to generate lenacapavir-resistant mutations. Drug-resistant HIV strains have evolved mutations that alter their proteins in such a way as to evade drug binding. However, because lenacapavir and the nuclear pore proteins in the host cell share the Site 2 binding site, the virus is caught in an evolutionary catch-22. Mutations that weaken lenacapavir binding run the risk of disrupting the host protein interactions that the virus needs to replicate. The same changes that might confer resistance to our drug could very well cripple HIV’s ability to replicate.
Courtesy of Gilead Sciences
Lenacapavir’s success sets an encouraging precedent for new ways of treating other viral infections. We showed that a small-molecule drug can meaningfully alter protein–protein interactions to disrupt a viral capsid’s assembly and disassembly. The success of this approach for combating HIV may someday lead to the discovery of novel drugs targeting the capsids of other viruses. All viruses known to infect humans have viral capsids, and those capsids likely play multiple essential roles in the viruses’ life cycles. Scientists are already trying to develop drugs that target the capsids of other viruses, including the hepatitis B virus and the dengue virus.
The near-term implications of lenacapavir for HIV treatment and prevention are, of course, the most exciting. By finding a drug that blocks a new target, we can successfully treat infections caused by multidrug-resistant HIV strains. Thus, lenacapavir expands the number of people who can be treated effectively.
But it is lenacapavir’s ability to prevent HIV infections that might truly change the course of the global HIV epidemic. Although the discovery of anti-HIV drugs that could prevent infection was monumental, the success of pre-exposure prophylaxis has been limited by the need for compliance. In the developed world, compliance is mostly controlled by the person on pre-exposure prophylaxis. However, in parts of the developing world, some of which have very high infection rates (up to one in five adults in some areas), a number of external factors can lead to people missing doses.
We showed that a small-molecule drug can meaningfully alter protein–protein interactions to disrupt a viral capsid’s assembly and disassembly.
Breakdowns in pharmaceutical supply chains can lead to precarious access to antiretroviral drugs. Also, in many areas, there can be profound stigma and discrimination attached to the use of anti-HIV drugs. We expect that the twice-yearly dosing of lenacapavir will partially offset some of the adherence, access, and stigma issues currently associated with the need to consistently take daily pre-exposure prophylaxis pills.
Lenacapavir’s promise led the World Health Organization in July to release guidelines that recommend use of the drug for preventing HIV infections. But lenacapavir’s value in preventing new infections will ultimately depend on how many at-risk individuals use it. And at the moment, there is a huge discrepancy between the number of people using pre-exposure prophylaxis and the number who could benefit from the intervention. For example, in the United States in 2022, only 36 percent of those who could benefit from pre-exposure prophylaxis were taking it, according to the U.S. Centers for Disease Control and Prevention. This discrepancy exists everywhere in the world, but it is perhaps most concerning in the resource-limited regions of sub-Saharan Africa where HIV prevalence is highest and where logistical and economic hurdles prevent anti-HIV drugs from getting into the hands of the people who most need them. These issues have been further complicated by the recent closure by the Trump administration of the U.S. Agency for International Development and by disruptions in the President’s Emergency Plan for AIDS Relief.
Overcoming these logistical, economic, and political hurdles will be important so that people across the world can get access to this powerful new drug. It would be a triumphant result of an almost two-decade search for a molecule capable of crippling the HIV capsid.
Click "American Scientist" to access home page

American Scientist Comments and Discussion
To discuss our articles or comment on them, please share them and tag American Scientist on social media platforms. Here are links to our profiles on Twitter, Facebook, and LinkedIn.
If we re-share your post, we will moderate comments/discussion following our comments policy.