When Averages Hide Individual Differences in Clinical Trials
By David Kent, Rodney Hayward
Analyzing the results of clinical trials to expose individual patients' risks might help doctors make better treatment decisions
Analyzing the results of clinical trials to expose individual patients' risks might help doctors make better treatment decisions

DOI: 10.1511/2007.63.60
In 1793, when yellow fever reached Philadelphia, killing hundreds of people, nobody knew its cause. But there was no shortage of theories. Based on these, a few desperate physicians devised increasingly radical treatments.
One of the most famous, concocted by the renowned Dr. Benjamin Rush, was the "Ten-and-Fifteen" purge, a combination of 10 grains of calomel, a mercury-based compound, and 15 grains of jalap (the poisonous root of a Mexican plant related to the morning glory). After administering this toxic brew, doctors then bled patients so profusely they often passed out. Miraculously, a few hearty patients survived both the sickness and its cure, reinforcing the doctors' belief in the value of their treatment.

Chris Dorley-Brown
The confusion of Rush and his contemporaries is understandable; given the natural variation in patient outcomes, it can be surprisingly difficult to tell whether a treatment is helping or harming patients. Things are no different today: Therapies that are useless or worse can still inspire great enthusiasm among physicians. But unlike Rush, modern physicians are at least somewhat protected from the human tendency to draw unwarranted conclusions by a statistical instrument called the clinical trial.
The most powerful form of trial, the randomized controlled clinical trial, was devised as a means of determining a treatment's effect when many other factors, including unknown ones, might affect patient outcomes. Over the past 50 years, large-scale human trials have paid rich dividends in lives saved and improved quality of life. In 1972, a Scottish epidemiologist named Archie Cochrane published a book urging physicians to follow the evidence of clinical trials in their practices. By the 1990s a group of doctors led by the Canadian physician David Sackett had coined the term "evidence-based medicine," and a movement was born. These physicians advocated that everyday treatment decisions be guided by the results of systematic reviews of clinical trials.
What could be more reasonable? And yet precisely because evidence-based medicine gives impersonal statistical data greater weight than clinical experience, it has met strong and at times emotional resistance from practicing physicians. Some see this resistance as a self-interested reaction, but we believe that it arises in part from a fundamental mismatch between the evidence provided by clinical trials and the needs of practicing doctors treating individual patients. Because many factors other than the treatment affect a patient's outcome, determining the best treatment for a particular patient is fundamentally different from determining which treatment is best on average.
We believe that changes in the way clinical trials are analyzed could offer at least a partial solution to this dilemma and yield the more detailed information doctors need to make better treatment decisions.
The clinical trial is a surprisingly recent invention. The first modern trial, conducted in 1947-48, showed that the newly discovered antibiotic streptomycin was more effective than the conventional treatment for tuberculosis. It was to be 15 years, however, before drugs routinely underwent clinical trials prior to being sold in the United States. In the late 1950s, severe birth defects were reported after the tranquilizer thalidomide was given to pregnant women. This tragedy spurred Congress to pass the Kefauver-Harris Drug Amendments of 1962, which finally forced manufacturers to prove that a new drug was both safe and effective.
The randomized controlled clinical trial became the standard means of providing this proof. The patients in this type of trial are assigned to one of two groups—the experimental group or the control group—and assessment of the outcome measure is typically blinded or masked (the assessing physician does not know whether patients received treatment). Randomization is the feature that gives trials the power to find the treatment's effect in the clutter of different patient risk profiles. If patients are randomly assigned to the experimental and control groups, risk factors should be equally distributed between the groups. Thus any difference in the aggregated outcomes of the two groups can be attributed to the effects of treatment.
Tom Dunne
The treatment-effect, as it is called, is typically a single number that summarizes the overall result of the trial. The treatment-effect can be expressed as the absolute risk reduction (the difference between the outcome rate in the experimental group and the outcome rate in the control group) or the relative risk reduction (the decrease in bad outcomes in the experimental group relative to the outcome rate in the control group). The absolute risk reduction is always a much smaller number than the relative risk reduction. For example, if a trial shows that a statin drug decreases the risk of heart attacks from 6 percent (the outcome rate in the control group) to 4 percent (the outcome rate in the experimental group), the absolute risk reduction is 2 percent and the relative risk reduction is 33 percent (the absolute risk reduction divided by the outcome rate in the control group).
Doctors are more likely to adopt a treatment when the treatment-effect is expressed using a larger, more impressive number, even though the information underlying calculations of absolute and relative risk is identical. Thus, trial sponsors (frequently pharmaceutical companies) typically emphasize the larger relative risk reduction. But whichever way treatment-effect is expressed, reporting a single number gives the misleading impression that the treatment-effect is a property of the drug rather than of the interaction between the drug and the complex risk-benefit profile of a particular group of patients. Consider what happens when sicker patients are enrolled and the rate of the problematic outcome in the trial goes up. If the relative risk reduction stays the same, the absolute benefit must get proportionally larger. This reflects our intuition that sicker patients have potentially more to gain from therapy.
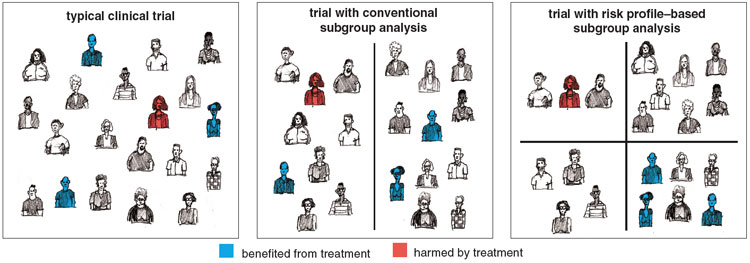
But when treatments have even a small risk of serious harm, the differences in treatment-effect may not just be a matter of degree. Indeed, some patients may benefit substantially from a treatment even when the overall results from a trial are negative. Or a treatment with benefit on average may be extremely unlikely to help most patients, while being more likely to harm than help some others. But unless the trial investigators analyze their data looking for these subgroups, the physician cannot know whether they exist.
Harm to a few was the problem lurking in the statistics of the landmark gusto study, which compared two thrombolytic (clot-busting) drugs for heart-attack victims. In the 1970s several drugs were found that could dissolve a clot and restore blood flow to heart muscle before it was irretrievably damaged. One of these was streptokinase. But in 1978 a Belgian scientist discovered that the cells lining blood vessels made an enzyme, tissue-type plasminogen activator, or t-PA, that also dissolved clots. In the early 1990s the biotechnology company Genentech, which had succeeded in genetically engineering this enzyme, and the National Institutes of Health sponsored a huge clinical trial of streptokinase and t-PA.

Tom Dunne
The trial showed that t-PA was considerably more effective than streptokinase, reducing the relative risk of death by about 15 percent. The newer drug was also much more expensive than streptokinase, but analysis showed that its benefits justified the additional expense. Following the gusto study, use of streptokinase declined dramatically, and it is now very rarely used for heart attacks in the U.S.
Of course, t-PA does not reduce every patient's risk by the same amount. Consider, for example, two patients who both qualify for thrombolytics. Estragon is 72 and diabetic. When he arrives at the emergency room by ambulance, he is experiencing severe chest pain and has a rapid pulse and low blood pressure. An electrocardiogram indicates a heart attack affecting a large and vital area of the heart muscle. Vladimir, 52, has stable vital signs and no chronic illnesses. He has come to the emergency room complaining of chest pressure. His electrocardiogram indicates that he has had a heart attack affecting only a small area of the heart muscle.
Given his condition, Estragon's mortality risk without thrombolytics would be about 25 percent, whereas Vladimir's would be close to 2 percent. Estragon is at such high risk of dying that the potential benefits of t-PA clearly outweigh any risks or costs associated with this agent. But it is not clear that t-PA would benefit Vladimir, who is highly likely to survive no matter which thrombolytic agent he receives. In fact, if Vladimir has high blood pressure or a history of stroke, both of which would increase his risk of intracranial bleeding, giving him the more potent t-PA might actually increasehis risk of dying (albeit only slightly).
In the gusto trial lower-risk patients like Vladimir were much more common than higher-risk patients like Estragon. When we re-analyzed the gusto results using mathematical models that estimated the risk of death based on patient characteristics, we discovered that t-PA primarily benefited a subgroup of high-risk patients. The highest-risk quartile of patients accounted for most of the outcomes that gave t-PA the edge over streptokinase. Paradoxically, even though the overall results of the trial suggest that t-PA is better and clearly worth the extra risks and costs, the benefits for the typical patient in the trial are less, and the trade-offs less clear.
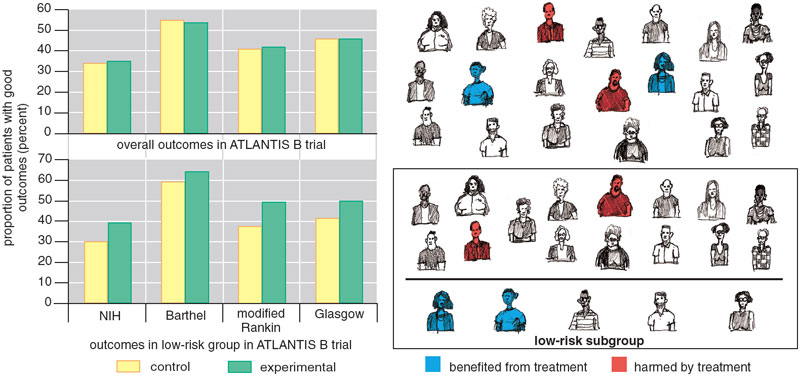
Summarizing trial results may exaggerate the benefit of treatment for some patients, but the reverse is also possible: A negative overall result can hide significant benefit to some patients. Consider, for example, the ATLANTIS B trial, undertaken in the late 1990s. This trial tested the efficacy of t-PA in treating strokes instead of heart attacks. Strokes are trickier than heart attacks because the thrombolytics must be given much sooner (within 3 rather than 12 hours) and the risk of thrombolytic-related intracranial hemorrhage is much greater (probably 6 or 7 percent instead of 1 percent).
Tom Dunne
Earlier clinical trials had shown that t-PA did not yield any overall benefit if it was administered more than three hours after the patient first had symptoms of a stroke. This short window of opportunity meant t-PA was given to fewer than 5 percent of stroke patients. To re-test the treatment window, ATLANTIS B enrolled patients arriving for treatment between three and five hours after the onset of symptoms of a stroke. The trial demonstrated no overall benefit for t-PA (treated patients were no more likely than those who received a placebo to recover normal or near-normal function). Moreover, as mentioned above, treatment with t-PA substantially increased their risk of intracranial hemorrhage.
Physicians looking only at the average result of this trial would be understandably discouraged by the lack of benefit and the increased risk of harm. But the trial showed that t-PA and placebo were essentially equivalent. The fact that some patients given t-PA were harmed by it implies others must have benefited from it. We hypothesized that if patients at lower risk of intracranial hemorrhage could be identified and t-PA given only to them, the treatment-effect might be different. When we used a risk model derived from independent data to divide the ATLANTIS B patients into thirds, we found that the third of the patient population at the lowest risk of thrombolytic-related hemorrhage actually did better with t-PA—even though they were treated outside the approved time window.
The paradoxical results of the gusto and ATLANTIS B trials arise from underlying variation in the baseline risks of these populations. For gusto, variation in the degree of benefit was due mostly to large variation in the risk of the outcome (death). For ATLANTIS B, it was attributable to variation in the risk of treatment-related harm.
John Ioannidis and Joseph Lau, our colleagues at the University of Ioannina in Greece and Tufts-New England Medical Center respectively, have advocated measuring the degree of variation in outcome risk in a trial by comparing the outcome rate in the quarter of patients with the lowest risk score to the outcome rate in the quarter with the highest risk score. In the gusto trial, the mortality rate in the highest-risk quartile is nearly 10 times higher than that in the lowest risk quartile.
Tom Dunne
This degree of variation may seem high, but it is not extreme by any means. Looking at trials of treatments for HIV infection, Ioannidis and Lau found examples where the outcome rate in the high-risk group was more than 50 times higher than that of the low-risk group. And when we looked at trials testing blood-pressure medicine for chronic kidney disease, we found similar ratios: Outcome rates were less than 1 percent in the low-risk patients and more than 30 percent in high-risk patients.
It should be obvious that when there is this degree of variation, one should not expect similar risk-benefit trade-offs in high and low risk groups. The high degree of variation arises because trials frequently enroll many patients with a negligible risk for the outcome even in the absence of treatment. For such patients, therapies associated with even a modest risk of treatment-related adverse effects will be causing net harm.
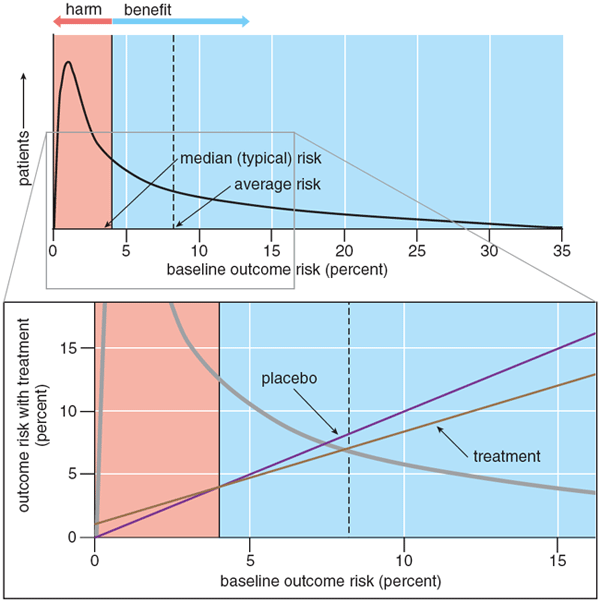
Not only is there considerable variation in risk, but it also appears that the baseline risk is not always distributed normally, in bell-curve fashion. If risk were distributed normally, then the overall trial result would at least reflect the outcome risk and treatment-effect in the typical patient. But in fact the gusto distribution, where many patients are at low risk and a few patients at high risk, might be more typical.
There are several reasons why risks might be highly skewed. One of them is something called the floor effect. Since there is no such thing as a negative risk, people with high outcome risks cannot be balanced out by people with risks less than zero. Another is that risk factors are not randomly distributed but instead clump: The presence of one risk factor often increases the likelihood of having others. When risks are skewed, the typical outcome risk may be different from the average outcome risk, and therefore the overall treatment-effect might not reflect the benefit to even the typical patient in the trial (Figure 5).
Many clinical trials include some attempt to explore differences in treatment-effect among the enrolled patients. But these analyses almost always focus on one attribute or risk factor at a time. For example, they might compare outcomes in men and women or in patients with and without hypertension. But one-variable-at-a-time subgroup analyses are not likely to yield meaningful information. For one thing, so many different variables can potentially influence the response to therapy and the likelihood of an outcome that if separate subgroup comparisons are made, chance alone will ensure that some subgroups show differences in treatment-effect. The hazards of "false positive" subgroup analysis from multiple comparisons were amusingly demonstrated using data from the isis-2 trial, which looked at the effect of aspirin in patients with heart attacks. Post-hoc subgroup analysis showed that aspirin did not lower mortality in heart-attack patients born under the signs of Libra and Gemini, but did in those born under other signs.
An equally important and less-well-appreciated reason that one-variable-at-a-time analysis is not effective is that a patient's outcome can be affected by many factors simultaneously. Since risks affect outcomes cumulatively, outcome differences between groups that differ by just a single risk factor tend to be relatively small. On the other hand, large outcome differences are found in analyses that compare subjects with many risk factors to subjects with none or few. Even if the experimenters pick a relatively strong risk factor, a single factor is unlikely to reliably discriminate between those who are at greatly different risks for the outcome and who therefore have widely diverging risk-benefit trade-offs from treatment.
We believe that to be truly useful clinical trials must routinely include analyses that combine risk factors into risk scores or indices. Risk models for a wide variety of diseases can be found in the literature, although they have not yet been exploited to analyze clinical-trial results.
To demonstrate that our gusto and ATLANTIS B examples aren't anomalous and that multifactor analyses are by nature more powerful than single-variable analyses, we ran computer simulations of hypothetical clinical trials. We then grouped patients according to the presence or absence of one risk factor or according to a risk score based on their count of risk factors. When we looked at the outcomes of these simulated patients, single-factor subgroup analyses proved statistically weak; it was unlikely such analyses would reveal real and often large differences in the treatment-effect. Subgroup analyses using multiple factors, on the other hand, were extremely powerful; under typical circumstances, these analyses would reveal important differences in the treatment-effect in different risk groups. (See "Finding Answers for Vladimir and Estragon," sidebar at end of article.)
The risk scores in our simulation examined only one dimensionof risk: the risk of having the outcome of interest. Important variation in this baseline risk is so common that analysis across this dimension should be routine. But the importance of other dimensions of risk should be explored in certain cases.
For treatments with a particularly high rate of serious adverse events, such as thrombolytics for stroke, scores for the risk of treatment-related harm may be helpful in discriminating patients likely or unlikely to benefit (as in our ATLANTIS B analysis). In some cases, there might be reason to examine characteristics that affect the relative responsiveness to therapy, such as time-to-treatment for thrombolytics or other emergency therapies—although, in this dimension, it might be hard to combine such characteristics into a score. Lastly, particularly for chronic diseases being treated over time in older, sicker populations, competing risks (the risk of succumbing to an illness not related to the treatment) could also give rise to differences in treatment-effect.
In any case, what is most important is that the myriad individual risk factors can be summarized into just a few risk dimensions, which are much more powerful than the individual variables in sorting patients into those likely and unlikely to benefit.
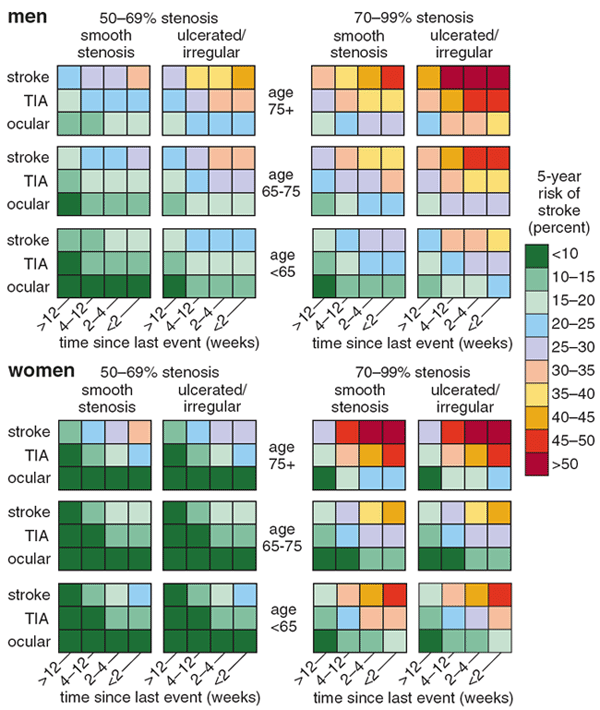
In 1999 Peter Rothwell of the Radcliffe Infirmary in Oxford, England, and Charles Warlow of Western General Hospital in Edinburgh, Scotland, published a landmark reanalysis that vividly shows how risk-benefit stratification can improve our understanding of a clinical trial. The trial, the European Carotid Surgery Trial (ecst), was designed to test whether patients who had recent warning signs or symptoms of a stroke benefited from carotid endarterectomy, a surgical procedure to clear plaque from one of the major arteries that carry blood to the head and neck.

Tom Dunne
This trial was a good candidate for reanalysis because treatment could be harmful as well as beneficial; debris from the artery could break off during surgery and migrate to the brain, causing a stroke. Patients had varying baseline risks for having a stroke if they did not have surgery, and they also had varying baseline risks of suffering harm during surgery. What's more, the factors predicting a patient's baseline risk were different from those predicting his or her risk of stroke during surgery.
The ecst trial showed that if patients had severe or "tight" stenoses (narrowings of the carotid artery that reduced its diameter by 70 percent or more), they benefited from endarterectomy, which reduced their five-year absolute risk of suffering a stroke by an average 7 percent. According to the overall trial results, all symptomatic patients with this risk factor should undergo surgery. Rothwell and Warlow reanalyzed the results for this group of patients.
The scientists derived two models for patient risk (risk of future stroke if untreated and risk of stroke during surgery) from other data. They then used these models to divide the patients with tight stenoses into subgroups and looked at the outcomes in these subgroups. It turned out that among patients with tight stenoses, only 16 percent benefited from surgery. Those who benefited were at relatively high risk of stroke if not treated but at relatively low risk of stroke during surgery. The other 84 percent of the patients had nearly identical outcomes with or without surgery. Again, although the average outcome suggested patients benefited, the typical patient did not. Reanalysis showed that only one in five patients with tight stenoses was helped by surgery.
Consider, for example, Cesario and Viola. Four days ago, Cesario—who is 76 years old—suddenly, though temporarily, lost control of his right hand and the ability to talk. A cerebral angiogram (an x-ray image of the arteries that supply the brain) showed that his carotid artery was 90 percent blocked, and the plaque had a highly irregular border. Based on this, according to the ecst model, his risk of a stroke over the next five years is over 40 percent.
Viola, on the other hand, is 59 years old. More than three months ago, she experienced transient loss of vision in one eye (suggesting a clot in the vessels supplying her eye rather than her brain). She has had no symptoms since. Her carotid blockage was 70 percent, and the plaque was smooth. Based on Viola's characteristics, her risk of stroke in the next five years is less than 5 percent. Moreover, because Viola is female and has very high blood pressure, her risk of stroke from the surgery is higher than Cesario's. For Cesario the benefits are clear; for Viola, the risks of stroke from the surgery itself would outweigh the benefits.
Once the benefits of risk-stratified analysis are explained, they seem obvious—so obvious one would think this type of analysis would already be commonplace. Yet risk-stratified analyses are rarely done. In 2001, we reviewed 108 clinical trials reported in four major journals. At that time we found only one trial that used statistical methods similar to those we suggest, and we haven't noticed many since.
Admittedly there are still methodological and practical problems with risk stratification to be ironed out. In a situation where there are factors that affect the outcome risk, factors that affect treatment-related harm and other factors that affect the responsiveness of patients to therapy, it can be difficult to know how best to combine these different dimensions to appropriately stratify patients. Also, there is never just a single way to describe risk. Different risk models using different variables may be equally valid but place individual patients into different strata, yielding different treatment recommendations depending on which model or score is applied. It should be noted, however, that the ambiguity about what to do for some individual patients currently exists; it is merely obscured by our insistence that the average benefit applies to all.
Tom Dunne
Additionally, moving risk stratification into the clinic will mean developing a usable set of tools for doctors to predict and communicate risk—and overcoming barriers to their adoption. Such tools might come in the form of charts, such as that shown in Figure 7, or in the form of informatics tools, such as the electrocardiograph-based predictive instruments developed by our coworker Harry Selker and other colleagues at Tufts-New England Medical Center (and used in our gusto reanalysis).
Finally, it must be recognized that there are considerable disincentives to taking this approach. What we call "individualized therapy" the pharmaceutical companies that sponsor most drug trials might call "market segmentation." If a trial results in a recommendation that all patients be treated, why look further and perhaps discover that only a subgroup of the patients is really benefiting? Indeed, the only robust risk-stratified analysis we found in our literature survey was done for a trial that showed no benefit overall but demonstrated beneficial effects in higher-risk patients.
Given these impediments, risk stratification, like the clinical trial itself, might not be widely adopted until regulatory agencies require it as part of the drug-approval process. To our knowledge the FDA has linked drug approval to a risk score only once. The prowess trial, published in 2001, showed that a new drug, drotrecogin, reduced mortality by 6.1 percent in patients with sepsis, organ failure caused by blood infection. Drotrecogin is a genetically engineered version of a protein normally found in the body that reduces clotting and inflammation. Because the drug is extremely expensive (about $7,000 per patient), the FDA advisory committee required that a risk-stratified analysis be performed on the prowess results.
When the patients were stratified according to the apache ii model (a well-known risk model), it turned out that the half of patients with lower apache scores fared no better with the agent, whereas the higher-risk patients benefited much more than the overall result suggested they would. The FDA approved drotrecogin only for these high-risk patients. Perhaps because they believed their overall results more than the risk-stratified results, the makers of drotrecogin then ran a second clinical trial limited to the lower-risk patients. This trial (the address trial) confirmed that drotrecogin does not benefit low-risk patients and may instead cause serious complications.
Because risk-based subgroup analyses are so rare, it is impossible to know how often this kind of clinically important variation in benefit goes undetected and leads harmfully to over- or under-treatment. One might say that the conventional approach to reporting overall results of clinical trials consigns us to an impoverished perspective similar to that described in Edwin Abbott's 19th-century science-fiction novella Flatland. In Flatland, characters inhabit a two-dimensional plane and perceive objects only if they intersect this plane; the world of three dimensions is unfathomable. In our medical Flatland, all the rich data from a trial is flattened into a single effect; a therapy either works or it doesn't. This binary outcome seems useful, since it conforms well to the binary decisions doctors must make: to treat or not treat. But the treatment decision is easy only because it's fitted to the average patient, not to real individuals. Analyzing and presenting clinical trial results across dimensions of risk can provide us with a more flexible, multidimensional evidence base for treating actual, not average, patients.
Click "American Scientist" to access home page

American Scientist Comments and Discussion
To discuss our articles or comment on them, please share them and tag American Scientist on social media platforms. Here are links to our profiles on Twitter, Facebook, and LinkedIn.
If we re-share your post, we will moderate comments/discussion following our comments policy.