The Life and Possible Death of the Great Asian Monsoon
By Sunil Amrith
The imperious storms have ruled India for centuries. Already unstable, what happens if they shift fundamentally?
The imperious storms have ruled India for centuries. Already unstable, what happens if they shift fundamentally?

These fat konrai [cassia] trees
are gullible:
the season of rains
that he spoke of
when he went through the stones
of the desert
is not yet here
though these trees
mistaking the untimely rains
have put out
their long arrangements of
flowers
on the twigs
as if for a proper monsoon.
The long wait for the rains suffuses many works of classical Indian literature. In these verses from The Interior Landscape: Classical Tamil Love Poems (1967), the poet and scholar A. K. Ramanujan evokes a sense of the monsoon’s illusions. The cassia trees are “gullible,” as people are prone to be. They mistake nature’s signs. They hope in portents of rain that prove false.
“As if for a proper monsoon”—but who knows what a “proper monsoon” looks like anymore? Over the past few decades, South Asian monsoons have swung between greater extremes of wet and dry. A monsoon’s mechanisms are so complex they resist modeling. We can send crafts to other planets, but no one can predict how much rain the monsoon will bring. Accelerating climate change brings even more uncertainty. If a fundamental shift in the patterns of the monsoon comes, it will devastate the livelihoods of hundreds of millions of people.
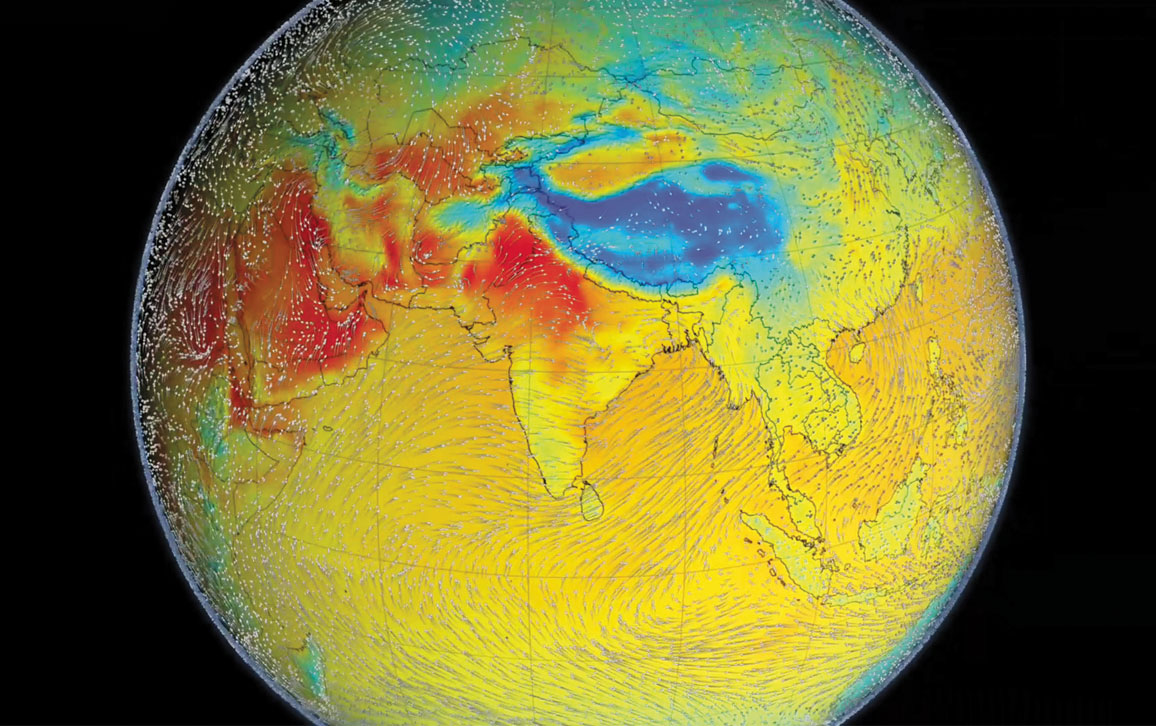
The word monsoon first appeared in English in the late-16th century, derived from the Portuguese monção, which comes from the Arabic mawsim (for season). Mawsim also provides the word for season (mausam) in Urdu and Hindi. In its simplest definition, it is a weather system of regularly reversing winds, one wet and one dry season. There are many monsoon systems around the world, but the South Asian monsoon is the greatest in scale and consequence. The Indian subcontinent constitutes the core of the monsoon system because of the geological history that has left India at the edge of the Eurasian landmass, which dominates the Northern Hemisphere. There India sits, facing the watery expanse of the Southern Hemisphere.
As the Asian continent heats up each spring, the warming air above it rises. Cooler, moist ocean air moves in to take its place. The monsoon winds blow from the southwest, curved by Earth’s rotation so that they double back to grip India from both the Arabian Sea, in the southwest, and the Bay of Bengal, in the southeast. The air sweeping in from the ocean contains vast stores of solar energy in the form of evaporated water. The vapor condenses and is released as rain. It is an extraordinary and powerful system. But it is not immortal or impervious to change.

Associated Press
The Himalayas are a crucial part of the system. The elevation of the Tibetan plateau leads it to warm rapidly, and so drives the differences of pressure and temperature that power the monsoon system. But the mountains themselves act as a colossal barrier to the winds, concentrating the monsoon rains to the south, along the Indo-Gangetic Plain.
More than 70 percent of total rainfall in South Asia occurs during just three months each year, between June and September. Within that period, rainfall is not consistent: It is compressed into a total of just 100 hours of torrential rain, spread across the summer months. Despite advances in irrigation, 60 percent of Indian agriculture remains rain fed, and agriculture employs around 60 percent of India’s population. No comparable number of human beings anywhere in the world depend on such seasonal rainfall.
Both before and after independence, the imperious power of the monsoon troubled India’s rulers. In the first decade of the 20th century, the finance minister in the imperial government declared that “every budget is a gamble on the rains”—a statement that is still quoted regularly in the Indian media. In the late 1960s, India’s prime minister Indira Gandhi said, “For us in India, scarcity is only a missed monsoon away.” The foreboding remains. In a lecture at Harvard University in 2017, the environmental activist Sunita Narain declared that “India’s finance minister is the monsoon.” This sense of a battle against enormous natural forces has inspired an enduring debate in India, among those who believe that science and technology could conquer nature, and those, like Narain, who advocate greater respect for nature’s limits.
The study of the monsoon arose from practical, commercial concerns. Seafarers mastered the winds that made possible their ocean crossings, and sailors also needed to understand the storms that menaced them. Knowledge of the monsoon was valuable and moved across cultures. The 14thcentury mariner Ahmad ibn Mājid provided the earliest written account of the monsoon in his Kitab al-Fawa’id (Book of Lessons on the Foundation of the Sea and Navigation).
By the 17th century, ships from the English and Dutch East India companies followed codified sailing directions on their voyages east. The logbooks from these voyages generated a vast store of weather data. In the 1840s, Henry Piddington, a retired ship’s captain and president of the Marine Courts of Calcutta, coined the word cyclone to describe the Bay of Bengal’s characteristic storms, driven by “circular or highly curved winds.” He derived it “from the Greek kukloma (which signifies among other things the coil of a snake).”
One such cyclone provided the catalyst for the development of meteorology in India. In October 1864, a “cyclone of unparalleled fury” struck Calcutta and the coastal districts of Bengal. The “rivers raged and tossed like a sea” and left the city “in ruins.” “Far as the eye can see,” wrote a British correspondent, “there is unbroken waste and gloom.” The destructiveness of the cyclone prompted the development of the Indian meteorological service, established in 1875. Its first director, Henry Blanford, had trained as a geologist but found himself drawn to the monsoon’s mysteries.
Greg Shirah, Horace/GSFC, Alex Kekesi/GST
That same decade, the 1870s, a series of famines provided a brutal reminder of India’s dependence on rainfall. They also brought to light the interconnectedness of cycles of drought around the world. Contemporary observers saw that India, northern China, Java, Egypt, and Brazil’s northeast all suffered simultaneously. It would take almost a century to work out why. Historian Mike Davis, emeritus of the University of California, Riverside, has called the famines “late Victorian Holocausts.” Far from being a “natural” disaster, he argued in 2001, the millions who died in the famines of the 1870s, and again in the 1890s, were “murdered . . . by the theological application of the sacred principles of Smith, Bentham and Mill.” Yes, the rains failed on a colossal scale, but what turned drought into disaster was imperial policy. Over the long term, British rule undermined the resilience of rural communities as they dragged India into modern capitalism. Over the short term, British rule made things worse by denying relief to starving people in the name of the integrity of “free” markets.
But even those who were most critical of the colonial government’s response to the famines concluded that India’s dependence on the rains was debilitating. The judge, social reformer, and economist Mahadev Govind Ranade (1842–1901) wrote in 1899 that “the last margin has been reached, and millions die or starve when a single monsoon fails.” The one thing that the colonial government and its Indian and British critics shared, from this point on, was an obsession with irrigation. Perennial irrigation, they believed, would do more than anything else to mitigate the vagaries of the monsoon. Their quest would transform the landscape and hydrology of South Asia.
By the end of the 19th century, forecasting was the tantalizing prospect before India’s meteorologists. In 1882, Blanford produced his first monsoon forecasts. They were tentative. To keep it manageable, Blanford focused on just one indicator: He was convinced that there was an inverse relationship between the quantity of snowfall in the Himalayas and the strength of a monsoon season to come. He predicted that less snow on the mountain peaks would lead the continent to warm faster in the spring and summer, strengthening monsoon circulation. His first attempts proved broadly accurate. But Blanford and his colleagues already suspected that the monsoon operated on a bigger scale.
The unveiling of those oceanic connections was the work of one of the most important climate scientists of the 20th century, Sir Gilbert Walker. He was a brilliant mathematician and a modest man. Walker’s career took him from Trinity College, Cambridge—where he studied the trajectory of boomerangs in flight across the lawns—to India’s weather service, which he directed from 1904 to 1924. He was an ornithologist and a flautist, as well as a master statistician.
We are left with a bitter irony. Through a cascade of unintended consequences, many of the measures taken to secure India against the vagaries of the monsoon have destabilized the monsoon itself.
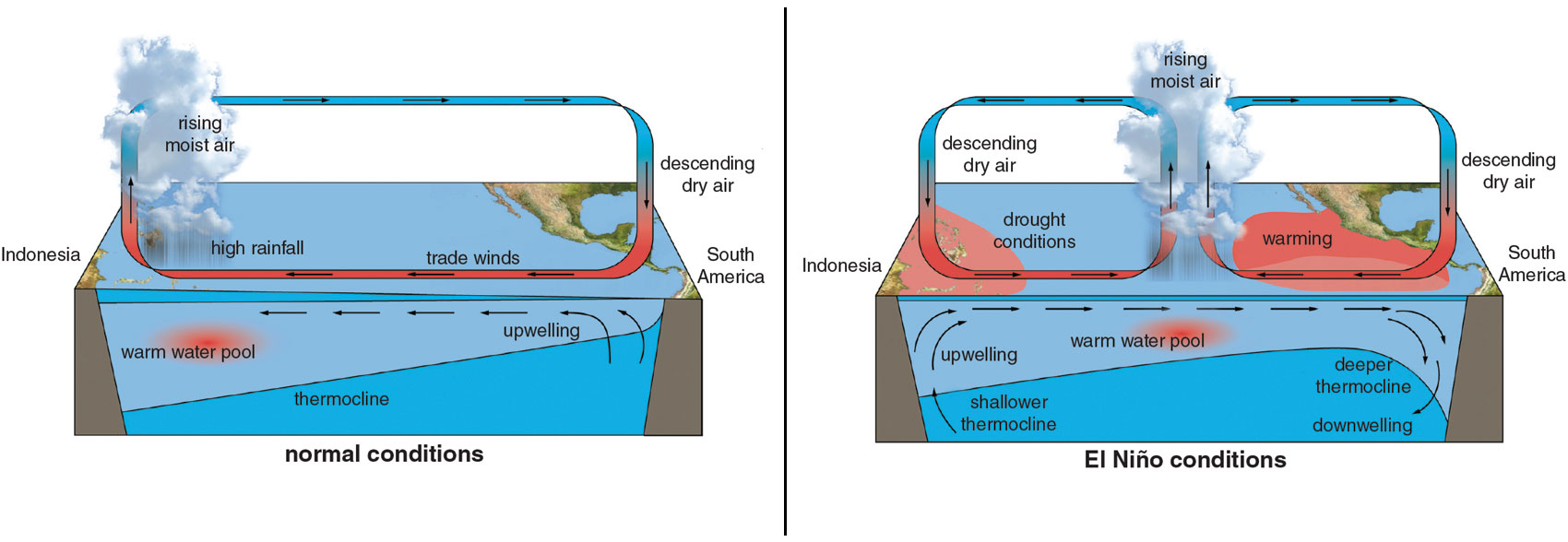
Walker took an empirical approach to understanding the monsoon. He relied on an army of Indian staff—a vast “human computer” led by Rai Bahadur Hem Raj, whose work and skill Walker acknowledged. Walker’s team acquired a quantity of data that, even just a generation earlier, would have been impossible. The numbers told a new story: They suggested that the monsoon system was planetary in its reach. The strength of the monsoon appeared to correlate with reversals in the pressure gradient across the Pacific Ocean. Walker called this phenomenon the Southern Oscillation.
Walker sought correlations in time as well as in space. He saw what he called foreshadowings of dearth or plenty in India as far away as Zanzibar or the Aleutian Islands in Alaska, and noted the lags of a season or two in the relationships he discovered. But the causal forces at work eluded him.
Forty years later, Walker’s insights would inspire Jacob Bjerknes, a meteorologist at the University of California, Los Angeles, to identify a quasiperiodic warming of sea-surface temperatures in the eastern Pacific Ocean, off the coast of Peru, with effects that reverberate throughout the world’s climate. Keeping with the term that local fishing communities used, Bjerknes called it El Niño. We know now that many of the worst droughts in Asian history correspond to intensive El Niño events.
The scale of the monsoon system exists far beyond human intervention. If technology could intervene, it would be on the landscape, in the form of infrastructure. By the early 20th century, engineers around the world were confident that they could neutralize the risk of climatic variability by constructing dams that would fuse water storage, flood control, irrigation, and power generation. India was no exception. In the 1930s, as they envisaged independence, many Indian scientists and politicians looked to the hydraulic achievements of the Tennessee Valley Authority, as well as to the Soviet Union, as a model.
© ESA/ATG medialab
At the time of independence, and in the aftermath of the brutal partition that divided India from Pakistan, large water projects promised to even out the fluctuations of a monsoon climate. They would boost food production in a part of the world where the memory of famine still stung, and where the partition left both new countries feeling stripped of valuable agricultural land. When he surveyed the Bhakra Nangal Dam in Punjab in 1956, India’s first prime minister, Jawaharlal Nehru, declared, “These are the new temples of India, where I worship.” Public information films extolled India’s liberation from an age-old struggle for water.
In the 1960s, this heady confidence of the early post-independence years received a blow. For two successive years, in 1965 and 1966, large parts of India suffered from monsoon failure. In the Indian state of Bihar, the government admitted that harvest failure threatened famine. Following the death of Nehru at the end of May 1964, the drought coincided with India’s first major political transition after independence. Food shortages pushed the country deeper into dependence on aid from the United States. “How helplessly we are at the mercy of the elements,” lamented a newspaper editorial in 1965.
As India received ever more U.S. aid, the administration of president Lyndon B. Johnson grew more interested in the monsoon. Johnson himself wrote in his memoirs that, overseeing grain shipments to India, he came to know “exactly where the rain fell and where it failed to fall in India.” As historian Kristine Harper of Florida State University showed in her 2017 book Make It Rain, in 1967 the U.S. government was involved in a secret and outlandish project to mitigate drought in Bihar using silver iodide to force precipitation. The project was a testing ground for more ambitious plans that the U.S. military had for Indochina. But “Project GROMET,” as it was known, failed and was quickly buried in the archives.
The crux of India’s response to the monsoon failures of the 1960s was to turn to the ocean of water underground. Large dams—however monumental, however many millions of people they evicted from their homes—simply could not provide enough water. The arrival of the tube well changed that decisively.
Tube wells are a humble technology, unlikely harbingers of a hydrological revolution. Driven by an electric pump, these wells consist of a long stainless steel tube that is bored into an underground aquifer (see “A Delta in Peril,” September–October 2019).
Tube wells domesticated the monsoon. They brought water year-round, and put it under the command of farmers who could afford the pumps, while strengthening their dominant position over those who could not. The contours of inequality in rural India assumed three dimensions, favoring those who had the capital to dig deepest in search of water.
A leap in the quantity of tube-well-irrigated land, combined with the high-yielding seed varieties that India imported from Mexico, produced what has come to be known as the green revolution. Just a decade after the droughts of the 1960s, India became self-sufficient in food, and with almost no expansion in the amount of land devoted to food production. With this change came an inversion of India’s map of water. The drier regions of India’s northwest and southeast propelled agricultural growth, thanks to groundwater irrigation. The monsoon-fed areas of India’s northeast, historically the centers of food production, now fell behind.
The much-celebrated successes of the green revolution created a sense among the Indian elite that the threat of an uncertain monsoon had receded. In a 1987 essay on the monsoon in Indian literature, the writer and newspaper editor Khushwant Singh cited a range of epics and poetry to show how deeply the monsoon had shaped Indian cultural sensibilities over hundreds of years. Singh concluded that, in recent decades, “India has taken enormous strides toward freeing herself from dependence on the vagaries of the monsoons.”
Joshua Stevens/MODIS data from NASA EOSDIS/LANCE and GIBS / Worldview
“There is no longer the same agony waiting through long summer months of searing heat to catch a glimpse of the first clouds,” he wrote. The monsoon had vanished from Indian literature; it “no longer stirs the imagination of the poet or the novelist with the same intensity it used to.”
At just this moment in the 1980s, climate scientists began to worry about the behavior of the monsoon. Late-20th-century breakthroughs in tropical meteorology brought new insights about the internal variability in the monsoon on multiple timescales, from the quasiperiodic impact of the El Niño Southern Oscillation to the intraseasonal variations attributed to the eastward moving equatorial fluctuation called the Madden–Julian Oscillation. With mounting evidence of anthropogenic climate change, meteorologists turned to the question of how planetary warming would affect the monsoon.
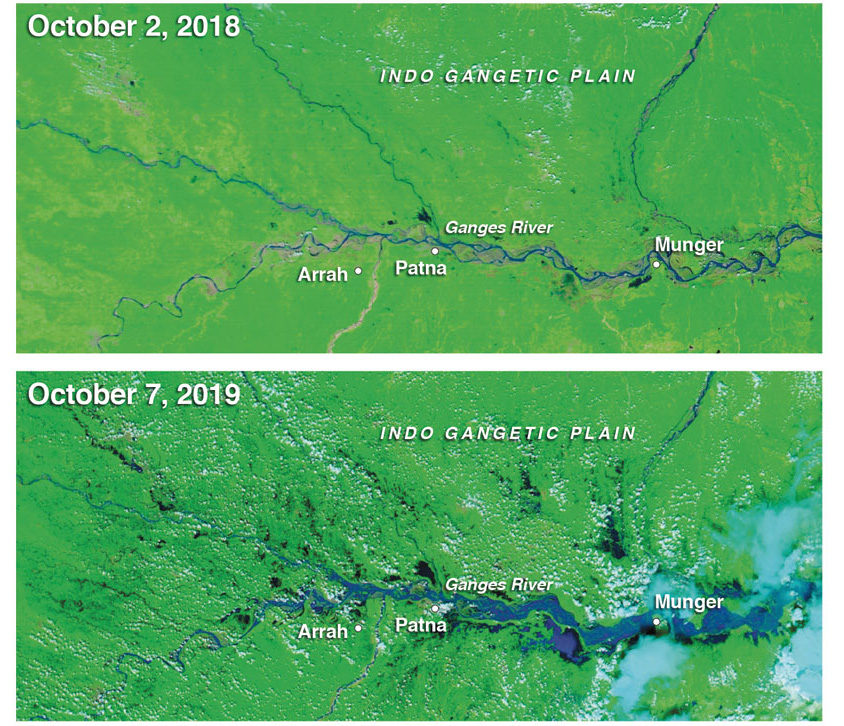
Although the monsoon is shaped by complex remote influences, it is also affected by transformations on a regional scale. Aerosol emissions—particulate matter from vehicle emissions, crop burning, and domestic cooking fires—are a major culprit (see “Dirty Rainbows,” From the Staff blog, April 13, 2018). The skies over India have the highest concentration of aerosols in the world, especially during the winter months when there is no rain to wash the skies clean. They appear on satellite images as a giant stain spreading across the Indian Ocean. Scientists have dubbed it the brown cloud. It poisons bodies. The death toll from indoor air pollution in India is estimated to be almost half a million people a year. Recent research has suggested that, by affecting the thermal contrast that drives the monsoon season, aerosol emissions have also contributed to a decline in monsoon rainfall. One recent study shows that remote aerosols, especially sulfates generated in East Asia, affect monsoon rainfall over South Asia.
Consider the dilemmas this raises. The brown cloud is a function of energy poverty in South Asia rather than excess. It is, at least in part, the result of the incomplete combustion of the cheapest, most polluting fuels—the only fuels accessible to the 240 million people in India who live without access to electricity. To reduce aerosol emissions would demand the more equitable distribution of electricity. And unless additional energy can be generated from renewable sources, we must accept that it would in turn increase India’s greenhouse gas emissions, mitigating the regional drivers of climate change while contributing to planetary warming. There are no easy solutions.

Visuals Stock/Alamy Stock Photo
Over the past 150 years, forest cover over most parts of Asia has declined dramatically. This change, too, affects the monsoon. The ecologists of the 19th century, known as desiccationists, who equated deforestation with drought, might have misunderstood the mechanisms at work. But it now seems that they were not wrong to believe that changes in the land could affect the rains. The intensification of agricultural production in India, and the use of more water for irrigation, have affected the moisture of the soil and its capacity to absorb or reflect heat. Crops reflect more solar radiation than forests, which tend to absorb it.
We are left with a bitter irony. Through a cascade of unintended consequences, so many of the measures taken to secure India against the vagaries of the monsoon—intensive irrigation, the planting of new crops—have destabilized the monsoon itself.
When all of these effects combine with the impact of global warming on the ocean and the atmosphere, the instabilities multiply. Far from counteracting the effect of greenhouse gases, the impact of aerosol emissions and land-use change complicates them. The monsoon is truly the wild card in many climate models.
The most dire scenario for the future behavior of the monsoon would come if global warming proves a “tipping element” that causes an abrupt shift towards a drier state. There is geological evidence of such rapid shifts in India’s rainfall regime in the past, as a result of natural rather than anthropogenic climate change. The irreversible loss of mass of the Greenland ice sheet might be such a forcing event, which could reverberate across the planet’s climate to affect monsoon circulation. Such a risk “remains speculative,” concludes one review of the science. But its implications are terrifying.
A changing monsoon affects every form of life that depends on it.
Faced with risks that are “inconceivably large,” writes acclaimed Indian author Amitav Ghosh in his 2016 book The Great Derangement: Climate Change and the Unthinkable, most societies are guided by “the inertia of habitual motion.” And given the gravity of any shift in the monsoon, discussion of these risks has been notably absent from mainstream Indian media. The recent march of farmers to Delhi, in conjunction with a special session of Parliament to discuss the plight of rural India, highlighted that climate change is not a distant prospect but a looming and potentially crushing reality for many.
Those who worry most about the monsoon are those who know it most intimately. A changing monsoon affects every form of life that depends on it. From the Gurukula Botanical Sanctuary in Wayanad in the Indian state of Kerala, the ecologist Suprabha Seshan and her colleagues cultivate endangered plants native to the ecosystem of the Western Ghats. “We refer to these plants as refugees,” she wrote in 2017; many have been rescued from areas where forests have already been cut down. The gardeners’ work depends on an intuitive knowledge of the weather.
But these patterns are changing. Meteorological research and local perceptions agree that the monsoon has grown more unpredictable. Local species are bewildered by the weather’s signals. Temperatures are too high for some mountain species to thrive, and rising temperatures bring new diseases.
“I worry,” Seshan concludes, “that the monsoon, with its moods and savage powers, might altogether cease.”
This article was adapted from an essay previously published in Aeon, aeon.co.
Click "American Scientist" to access home page

American Scientist Comments and Discussion
To discuss our articles or comment on them, please share them and tag American Scientist on social media platforms. Here are links to our profiles on Twitter, Facebook, and LinkedIn.
If we re-share your post, we will moderate comments/discussion following our comments policy.