
This Article From Issue
July-August 2022
Volume 110, Number 4
Page 208
An average human brain is anything but average: It contains roughly 100 billion neurons, linked together by at least 100 trillion synaptic connections, bathed in ever-shifting chemicals and reacting to ever-changing stimuli. Making sense of all that complexity might seem like an unattainable goal, but Hongkui Zeng is working on it. She has sought to break down the problem into comprehensible chunks, investigating activity patterns, genetics, and cell types in the brain. Zeng also started on a reduced scale, first studying fruit flies, then moving up to mouse brains. Now, as the director of the Allen Institute for Brain Science in Seattle, Zeng is going all in. She is a key player in the BRAIN (Brain Research Through Advancing Innovative Neurotechnologies) Initiative, a vast, interdisciplinary alliance created to map out exactly how the human brain functions—and what happens when it malfunctions. Special issue editor Corey S. Powell spoke with Zeng about how she is approaching the colossal challenge of understanding the human mind. This interview has been edited for length and clarity.

Erik Dinnel / Allen Institute
What is the BRAIN Initiative aiming to accomplish, and how does your work fit within that mission?
The primary goal of the BRAIN Initiative is to develop neurotechnologies that will facilitate the understanding of the circuit function of the brain and how it changes in diseases. In conjunction with that goal, the BRAIN Initiative is also addressing series of questions using those technologies. They’ve set up seven different priority areas, from cells to circuits, from manipulation to the development of computational series, to explain how the brain works.
The number one priority out of the seven is to understand the cellular composition and diversity of the brain—basically coming up with a catalog of cell types and understanding the relationships, structures, and functions of those cell types—as a foundation to understand the brain itself. To understand how a car works, you need a list of parts, and you need to know what each part is doing and how the parts work together to allow the car to run. Cell types are the parts list of the brain. The BRAIN Initiative established a BRAIN Initiative Cell Census Network [BICCN] that is tasked with generating the cell type catalog for the mammalian brain, including major species from mice and rodents, up to humans and nonhuman primates.
Ad Right
We at the Allen Institute have been interested in the cell type question from the very beginning. We generated a brain atlas by profiling all 20,000 genes in the mouse genome and mapping how they are distributed in the brain. We’ve been doing that in the human brain as well. We have already established multiple transcriptomic technology platforms that allow us to profile individual cells in the brain very efficiently.
You used the term “brain circuit,” which brings to mind a digital computer. To what degree is that a useful paradigm?
It is a very good analogy, comparing the brain to a computer. In a computer, individual chips, or units, are connected to each other in a wiring diagram in order to perform computations between those different individual units. The brain is a much more advanced computer. The individual cells and cell types are the units. But the cell types are much more flexible than a computer chip, and they are not all the same unit. They’re diverse. They have different properties. They can change and tune their properties. They don’t just perform linear functions. The wiring diagram of the brain is the interconnections between the cells—the neurons of the brain. That wiring diagram is extremely complex and specific, which allows the brain to compute in sophisticated ways that we don’t understand yet.
How do you break down a task—such as mapping the brain, which is so big and complicated—into tangible units?
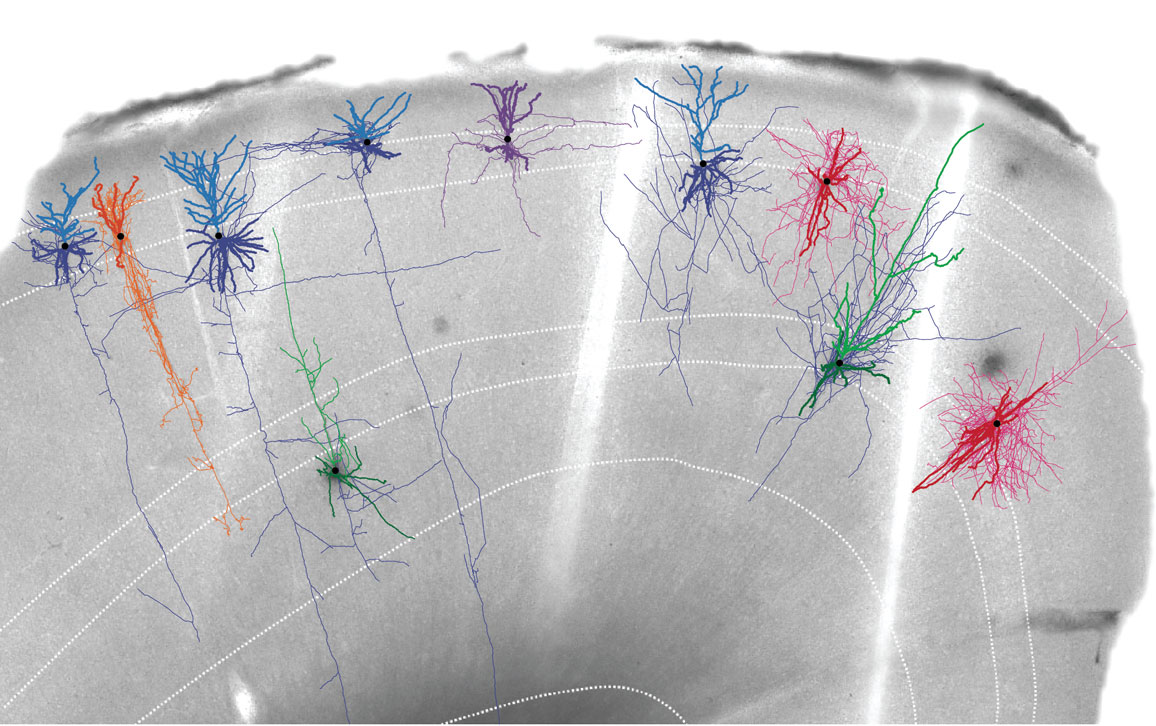
Fortunately, the brain is already broken down into units: the individual cells. What we need to do is characterize and measure the different properties of the cells.
We call the genes that a cell expresses transcriptomics. Transcription is the expression of the gene. If we obtain a transcriptome, it’s a collection of the expression level of all the genes in the genome. Then there’s the shape of the cell, the firing action, and the potential firing properties of the cells. There are other properties you can measure.
You do all these different quantitative measurements from the same cell, and then you can use clustering analysis to look for similarities in the properties between cells. When you use clustering analysis, you can identify types of cells. A cell type is a group of cells that have similar properties with each other, and different properties from other types. The complex issue here is that the cellular properties are very diverse. That’s why we’ve developed technologies to crack the cells in different ways.
Preferably you want to measure the different properties in the same cell so that you can correlate and understand the relationships between properties better. If you measure property A in cell 1 and then measure property B in cell 2, it’s very difficult to understand how properties A and B are related to each other, and how they’re related to cells 1 and 2. But if you measure A and B in 1, and A and B in 2, and then A and B in many other different cells, the cluster analysis will make sense and help you understand whether A and B characterize different types of cells, and whether A and B always go together. We call that multimodal analysis.
Another important feature is scalability. There are millions to billions of cells. A mouse brain contains 75 million cells. A human brain contains 82 billion cells. If you want to gain a systematic understanding—if you want to have a complete parts list—you have to profile millions of cells from the mouse brain. Choosing the right technology to scale up is very important. Currently, the most scalable approach is single-cell transcriptomics, which describes the entire repertoire of gene expression in a cell. Single-cell transcriptomics is a revolutionary technology developed within the past 10 years. It’s now being widely used to profile millions of cells.
How much of the cell categorization is qualitative and how much of it involves discrete, quantitative differences?
Everything is quantitative. You can assess qualitatively, but that is not good enough for an unbiased comparison between different kinds of cells. You always have to be quantitative.
Cell types are organized in a hierarchical manner. You can group cells into big categories—let’s say excitatory neurons and inhibitory neurons. They’re qualitatively different because they express two different neurotransmitters, but within each of those two different categories there will be many different types. Within each type, there could be many subtypes. So the difference between the types becomes smaller and more quantitative. The difference between subtypes within each type can be even more subtle, and sometimes it becomes a continuum.
At what point do you stop? How many categories do you find useful?
That’s something neuroscientists and biologists in general have been debating for a long time. It’s still an open question. You may have heard the debate between lumpers and splitters. Some people really want to know the details and just keep splitting groups into subtypes, whereas others—the lumpers—only care about major differences. Where should we stop? The major distinctions and major type levels are obvious, but as to where we stop, that comes from the multimodal analysis.
We know that single-cell transcriptomics is the most scalable approach. We can use that method to classify cells into types. However, when you get to the lower branches, it’s very hard to find out how many types there are. At that point, it is important to bring in additional features: the shape of the cell, the connections of the cell, and the physiological properties of the cell. Then we see how the different properties are covariant within a cell. When you introduce multimodal criteria, it helps to discretize, or it helps to prioritize the ambiguity between cells.
Of course, sometimes the process brings more ambiguity. Sometimes it brings less. But when it gets really confusing, then you say, “Let’s stop. We can’t divide these cells into types anymore.” But if there’s a point where you can clearly separate cells into groups based on multiple pieces of evidence, then you continue.
When you’re looking at the cell types by genomics, how do you mark the cells so that you know what you’re looking at?
When we do genomics or transcriptomics studies, we don’t need to mark the cells. The transcriptomics profile is already so rich. It has expression information on every gene. Once we cluster them, we can use marker genes—basically genes that tell apart the types. We use the characteristic genes that tell apart different clusters or types as the label markers.
Think of learning, disease, and cognition as players in an orchestra. Then you can begin to pick out how the players work together to make music.
The marker genes are the most differentially expressed genes. With clustering analysis, you group the cells, say, into two clusters. Then you compute the average gene expression level for every gene between the two clusters. You identify the discrete differences and find the genes with the biggest difference between the two types. Or, if you have many types, you identify the genes that are most unique, most differentially expressed in your cluster versus all the other clusters. Those are the cell-type marker genes.
When you go into other kinds of analysis, where you don’t have this rich information, you then need to label the cells. You use those marker genes to label. For example, you use the promoter of that marker gene to express a fluorescent protein, and you can decide that any cells labeled by this fluorescence belong to this transcriptomic type. Now you can measure its firing properties, its connections, its functions, things like that.
Allen Institute and Southeast University (Nanjing, China)
You alluded to three orders of magnitude in the jump from the mouse brain to the human brain, just in terms of cell numbers, never mind complexity. Where are you in laying the groundwork for doing the equivalent project on the human brain?
The groundwork would be on both the conceptual and technical levels. At the technical level, we have to scale up from what we do in the mouse. We probably don’t need to scale up proportionally, profiling a thousand times more cells in order to understand the human brain. But at least tenfold, maybe one-hundredfold.
The reason I say that we don’t need to do a thousandfold more is because of the conceptual advances we’re generating now in the mouse brain. We understand the organization, the basic parts list, how they’re distributed spatially, how they compose different systems of the brain. There’s the motor system, sensory, cognitive, memory, the innate behaviors. There are all these different parts of the brain. We now can provide the cellular composition for each part of the brain—the general network. That information can be carried over into the human brain, which helps us to develop a strategy to sample the different parts of the human brain for comparison with the mouse brain to see whether the cellular diversity is really a diversity difference, or just a numbers difference.
You go to some regions, you sample, and you find that actually the number of cell types in a homologous human region and mouse region doesn’t change much, even though the number of cells in those two regions changes a hundredfold. That just means there are more cells of a given type in a human brain compared to a mouse brain.
That’s a very important thing to find out, though—whether there are a thousand times more cell types in a human brain, or if it’s just more cells of each type. How does cell type scale up, and in what way? Having a comprehensive mouse map will help us to understand that question. We can analyze the complexity one region at a time and know what to look for.
Allen Institute
Did you run into any unexpected hurdles working on the mouse model that will help your process when you move on to the human brain?
Initially we didn’t know whether we’d be able to scale up efficiently. Maybe cost would be a hurdle. Whether there’s a technique that can give us sufficient resolution or not, and whether the technique would be cheap enough to allow us to profile many cells or not. There were hurdles at the beginning, but it’s amazing to see the technological progress over the past few years. Sequencing has become cheaper and cheaper. There are techniques that have emerged that allow us to scale up.
A technique that overcame one of the major hurdles is called spatial transcriptomics, or spatially resolved transcriptomics. My expectation is that our current single-cell transcriptomics will be replaced by spatial transcriptomics in the near future. With the previous single-cell transcriptomics, you have to dissociate the brain. You need to disrupt the brain tissue, add some proteinase or something, and really digest it and pull it apart. Then you isolate the single cells, sort them, put them in microfluid, and you get the transcriptomes of each cell. In that kind of process, you lose the spatial context of the cells completely. You don’t know which cell comes from where. But the spatial transcriptomics allows you to profile gene expression of brain sections in situ.
For spatial transcriptomics, you lay down the brain section on a barcoded array on a chip, and you allow the expressed genes—the transcripts—to be bound onto that barcoded array. That gives you not only what genes are expressed, but in which spots those genes are detected. It gives you both spatial information and gene expression information at the same time.
To create a brain atlas, you need to know not only what cells there are in the brain, but also where they are. A brain cell atlas without this spatial information is not a brain cell atlas. Coupling spatial and transcriptomic information together was a major hurdle for us, but there are techniques now that allow us to do that.
How do you put a map of cell connections and a map of cell types together to understand what’s actually going on in that brain?
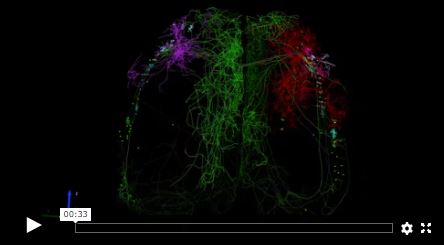
The idea is to understand how the cell types are wired together. For that, you need to label the different cell types. Then, using microscopy, you label different cell types, collect the imaging data, and trace. You literally trace individual neurons to find how they’re connected, where the synapses are on the neurons. And you derive, manually or computationally, the wiring diagram.
How do the principles of convergence science help you bring different types of expertise together to work on these problems?
We have to bring people with different expertise together—genomics people, neuroscientists, imaging and microscopy people, computational people. The major challenge is letting each of them understand what the others are doing. Very often, the problem has to be solved computationally. For example, the analysis of the genomics data as well as the analysis of the images—the light microscopy, and especially the electron microscopy images—takes a lot of computation. Especially for the electron microscopy images, there’s a lot of AI and machine learning involved in learning how to reconstruct faster. We’re dealing with really large-scale data sets now, and computation is critical.
How has your work deepened our understanding of the brain and brain disorders?
The work we’ve done in the mouse brain lays a much more detailed and comprehensive foundation for us to begin to understand the human brain and its normal functions, as well as changes in a disease situation. I emphasize the words detailed and comprehensive because you can’t look at a human brain and figure out exactly what’s happening with fuzzy observation techniques. By laying things out clearly, at a specific cell type level, it allows you to really dissect what is happening.
A lot of this knowledge can be translated into humans. Humans and mice, after all, are 90 percent similar to each other. Of course there are also areas that cannot be translated, but we can identify the similarities and the differences between a mouse brain and a human brain. And then, in terms of human diseases, you’re able to dissect which particular parts of the brain or parts of the pathways are changed, and what kind of changes happen, in a disease context. It gives you a much clearer, crisper picture than what we had before. We provide a detailed map to allow you to understand the variations of the brain.
Think of learning, disease, and cognition as players in an orchestra. Then you can begin to pick out how the players work together to make music. In cognition, which parts are activated? This group and this one are working together and generating this outcome, whereas that group is forming memories, and this change is related to some kind of disease. And you can begin to derive an active map. Instead of the static map that we initially have, you build activities, build dynamics, and build function into that map.
A podcast interview with the researcher:
American Scientist Comments and Discussion
To discuss our articles or comment on them, please share them and tag American Scientist on social media platforms. Here are links to our profiles on Twitter, Facebook, and LinkedIn.
If we re-share your post, we will moderate comments/discussion following our comments policy.