Talking Pictures
By Felice Frankel
Harvard astronomer Alyssa Goodman on the visual representations in her work
Harvard astronomer Alyssa Goodman on the visual representations in her work

DOI: 10.1511/2006.60.358
Alyssa Goodman, a friend and colleague, is professor of astronomy at Harvard University and director of Harvard's Initiative in Innovative Computing (http://iic.harvard.edu), with which I will become officially involved in the future. Alyssa and I try to grab the too-few available moments to discuss issues in the visual representation of science, respecting each other's differing perspectives. A distinguished research astronomer, Alyssa has a rare talent for knowing how to talk to audiences. This image is a slide from one of her talks.
F. F. Before we talk about this slide, can you tell us how you think about creating a slide—how you begin the process?
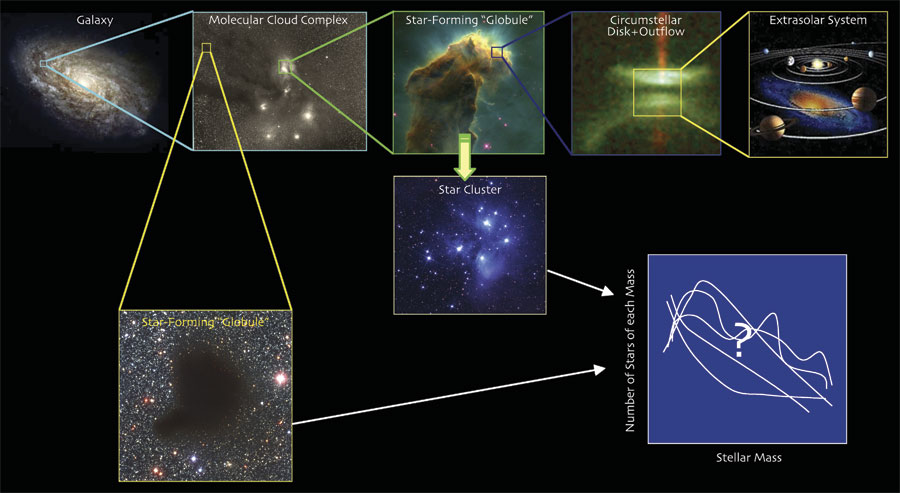
From upper left: molecular cloud complex, E. E. Barnard, 1907; Eagle Nebula, Jeff Hester and Paul Scowen, Arizona State University and NASA; circumstellar disk with jet, Hubble Space Telescope; artist's conception of extrasolar system in the making, source unknown; near-infrared composite image of "globule" B68, João Alves, European Space Observatory; star cluster, David Malin/Anglo-Australian Observatory.
A. G. I usually plan out a whole slide in my head, keeping the audience for it in mind, before I ever open my computer, much less PowerPoint. Often I do this planning while not sleeping at night, or in the shower. Then I try out the idea in PowerPoint, and I usually only add "fancy" features like animation when they explicitly enhance the point. The slide you've selected is an animated one. It begins with the largest-scale image, at top left, and then zooms in on each click to the next smallest scale, from right to left across the top row. The images of an individual star forming, and then of a cluster forming, are added for clarification later. And the individual "curves" in the plot of the mass function are flashed one at a time, automatically, in succession, to give the idea that the star-formation process could give a variety of results, even though in fact it mysteriously nearly always gives the same kind of distribution for stellar mass.
F. F. Where do you think most researchers make their biggest mistake when creating a slide for a talk?
A. G. TMI—"too much information"—usually in the form of too much text. Slide-making software should not be a way to put your notes up on a screen or to record everything you're going to say for posterity. I like to say "PowerPoint doesn't kill presentations, bullets do."
F. F. Would you say that creating your presentations is a means of clarifying your own ideas?
A. G. Absolutely. Whenever one is forced to organize information clearly, logically and concisely, the information inevitably becomes clearer to its organizer. This is especially true for presentations, where a presenter's time and an audience's attention span are always limited. I usually think very hard about who the audience is for any visual or slide. In the class on the "Art of Numbers" that I teach at Harvard, I am constantly telling the students to focus on two mantras: "who's the audience?" and "less is more."
[Information-design expert Edward] Tufte has criticized PowerPoint, in particular, for being "relentlessly sequential." For people like me, who like to skip around from idea to idea, depending on an audience's reaction, this can be really limiting. Nonetheless, I see two "bright-side" answers to Tufte's concern. First, being forced to come up with at least one semi-linear way to present your ideas can actually clarify the logic of one's thinking about the ideas being communicated. Second, judicious use of the "hyperlinking" features that programs like PowerPoint and Keynote make available can avoid the linearity constraint. I often put tiny icons on my slides that offer links to specific other slides that I may want to jump to at that point in a presentation. The new (and still a little buggy) "Presenter Tools" mode in PowerPoint also offers the same kind of random access to individual slides that acetate viewgraphs used to offer—only without the danger of dropping the stack on the floor!
Click "American Scientist" to access home page

American Scientist Comments and Discussion
To discuss our articles or comment on them, please share them and tag American Scientist on social media platforms. Here are links to our profiles on Twitter, Facebook, and LinkedIn.
If we re-share your post, we will moderate comments/discussion following our comments policy.