How to Fight Back Against Antibiotic Resistance
By Gautam Dantas, Morten O. A. Sommer
Mapping the exchange of genes between pathogens and nonpathogens offers new ways to understand and manage the spread of drug-resistant strains.
Mapping the exchange of genes between pathogens and nonpathogens offers new ways to understand and manage the spread of drug-resistant strains.

DOI: 10.1511/2014.106.42
Not so long ago, it seemed like the fight against infectious diseases was nearly won. The discovery of penicillin in 1929 gave clinicians their first weapon to combat common ailments like pneumonia, gonorrhea, and rheumatic fever. In the decades that followed, medical researchers discovered more than 150 other types of antibiotics. These widely hailed “wonder drugs” were so successful that U.S. Surgeon General William Stewart announced in 1967, “The time has come to close the book on infectious diseases.”
Science Source
Stewart and most of his contemporaries greatly underestimated the ability of bacterial pathogens to adapt to these life-saving medicines. Almost as soon as clinical use of penicillin began in 1946, the first drug-resistant pathogens appeared. During the golden age of antibiotic development (the 1940s to the 1960s), the spread of antibiotic resistance was balanced by the continued discovery and deployment of new classes of antibiotics. But starting in the 1970s, a dwindling interest and ability of the pharmaceutical industry to develop new antibiotics resulted in a 40-year period when virtually no new broad-spectrum classes of antibiotics were brought to the market. Instead, companies focused on modifying the chemical scaffolds of already approved classes of antibiotics.
During this innovation gap, bacterial evolution did not cease. Consequently, drugs that were previously effective in treating a broad spectrum of infectious bacteria are now useful for fewer and fewer infections. Certain bacteria, including strains of Escherichia coli and Klebsiella pneumonia, are now resistant to all major antibiotics— even carbapenems, which have long been the drug of last resort to treat afflictions such as lung infections. With dwindling treatment options, the mortality rate from those infections in the United States is approaching 50 percent. In effect, for some diseases we are now living in a post-antibiotic age.
According to a September 2013 report from the U.S. Centers for Disease Control and Prevention (CDC), treatment of antibiotic-resistant infections adds $35 billion in health care costs and 8 million hospital days per year in the United States. A recent drug-resistant Salmonella outbreak due to contaminated chicken meat was linked to nearly 300 illnesses across 18 states, sickening infants and nonagenarians alike. At least 23,000 Americans die each year from infections, many caused by the superbug methicillin-resistant Staphylococcus aureus (MRSA), because doctors have run out of drugs with which to treat them.
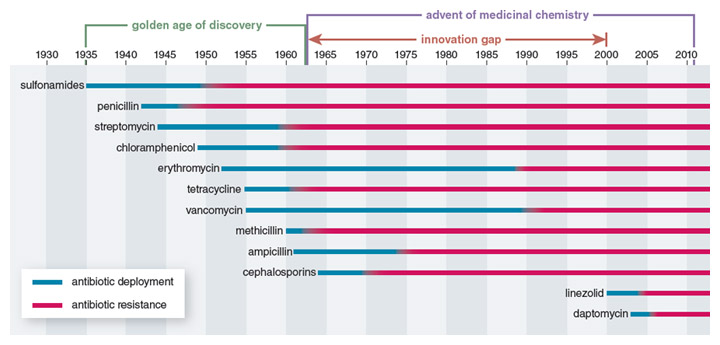
Illustration by Barbara Aulicino
Government agencies are belatedly considering incentives to support renewed antibiotic drug development, but these initiatives have not yet had a direct impact on the drug development pipeline. As a result the number of antibiotics approved by the Food and Drug Administration (FDA) hit a record low of one new antibiotic in the five-year period from 2008 to 2012, down from 16 new drugs in the years from 1983 to 1987. CDC Director Tom Frieden recently warned, “If we don’t act now, our medicine cabinet will be empty and we won’t have the antibiotics we need to save lives.” In reality, the development of new antibiotics is only part of the solution, as pathogens will inevitably develop resistance to even the most promising new compounds.
To save the era of antibiotics, scientists must figure out what it is about bacterial pathogens that makes resistance inevitable. By studying the suite of genes—collectively known as the resistome—that can turn a susceptible pathogen into a superbug, researchers may be able to uncover the Achilles heel of these multiple drug–resistant strains. Although most studies on drug resistance have focused on disease-causing pathogens, recent efforts by the two of us and by a number of our colleagues have shifted attention to the resistomes of nonpathogenic bacteria. Importantly, over the past decade advances in DNA sequencing have enabled us to explore the genomes of both pathogenic and non-pathogenic bacteria across a variety of different natural habitats.
This research has led to a growing understanding of how antibiotic resistance evolves in specific bacteria, as well as how it gets passed between different bacteria and between different environments. We are still a long way from Stewart’s old proclamation of victory, but the recent advances are helping frame new strategies to complement the 90-year-old paradigm of just trying to defeat resistant bacteria by discovering newer drugs. Understanding factors that influence resistome evolution and dissemination may both extend the life of current drugs and point toward new disease-fighting strategies.
There are two ways that pathogenic bacteria can develop antibiotic resistance: vertically, through the accumulation of genetic changes during the natural process of copying its genome, and horizontally, by swapping resistant genes from one microbe to another.
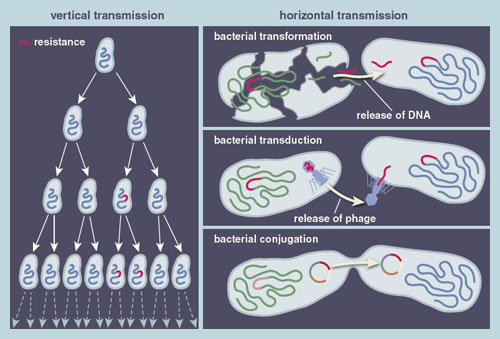
Illustration by Barbara Aulicino
Vertical transmission is the fundamental evolutionary process by which a cell can accumulate errors in its genome during replication, such that the resulting progeny differ genetically from their bacterial ancestors. The genome replication or copying error rate is rather low, so typically one in a thousand growing bacteria will introduce an error (called a mutation) into the genome. Not all of these mutations are advantageous, but about one in a billion will generate mutants that can grow faster or tolerate higher concentrations of antibiotics than their predecessors. When such bacterial mutants are exposed to antibiotics, those possessing antibiotic resistance genes will increase in prevalence to the point of taking over the entire population. Multiple cycles of such mutation and selection are often necessary to evolve high-level antibiotic resistance.
Genes that have evolved to confer antibiotic resistance to one type of bacteria can be transferred to another by a mechanism known as horizontal gene transfer. While some pathogens acquire resistance via vertical transmission, recent studies have suggested that horizontal transmission may be the dominant force behind growing antibiotic resistance. During horizontal gene transfer, antibiotic resistance genes catch a ride on mobile genetic elements that carry the genetic material between different cells. Mobile genetic elements can be linear or circular pieces of DNA, called plasmids, which are replicated by the cell along with its genome.
Understanding factors that influence resistome evolution and dissemination may both extend the life of current drugs and point toward new disease-fighting strategies.
These DNA fragments make their way into a new cell through three mechanisms: transformation, transduction, and conjugation. In transformation, bacterial cells scavenge DNA remnants from dead bacterial cells and integrate them into their own genome. In transduction, genetic material is transferred by bacteriophages (viruses that infect bacteria). Bacteriophages can insert their DNA into the genome of a host cell, where they are maintained for many generations before they pack up their DNA and leave to infect another cell. Along the way, the bacteriophage may coincidentally integrate a section of the bacterial host cell genome into the bacteriophage genome, enabling genetic material from one cell to hitchhike a ride to another cell on the bacteriophage.
The final way antibiotic resistance genes can be passed from one microbe to another is through conjugation, also known as bacterial sex. The discovery of this process, now thought to be the main mechanism of horizontal gene transfer, earned Joshua Lederberg the 1958 Nobel Prize in Physiology or Medicine. During conjugation, plasmids hijack the cellular machinery to create structures called pilli that protrude from the donor cell to penetrate the membrane of the recipient cells, enabling the transfer of the conjugative plasmid and all the functions it encodes. Many hospital-associated pathogens, including the carbapenem-resistant bacteria mentioned previously, harbor large conjugative plasmids that contain tens of resistance genes, offering the host cells immunity to virtually all antibiotics.
Recent large-scale efforts sequencing the genomes of many bacterial pathogens, by such groups as the Wellcome Trust Sanger Institute in the United Kingdom and the Broad Institute of MIT and Harvard in the United States, have added another complication to this story. These studies have shown that the genes conferring resistance toward antibiotics in pathogens can be acquired via horizontal gene transfer from another gene reservoir entirely, such as city soil, waste water, or processed meat. Accordingly, there is a substantial interest in broadly characterizing these gene reservoirs to improve our overall understanding of how swapping genes from one reservoir to another contributes to the evolution of antibiotic resistance in bacterial pathogens.
Just as there are a number of different ways for bacteria to acquire an antibiotic resistance gene, the genes themselves represent a number of different strategies to encode resistance. The genes that confer antibiotic resistance can be loosely separated into four groups, each with their own unique mechanism for combating antibiotic exposure.
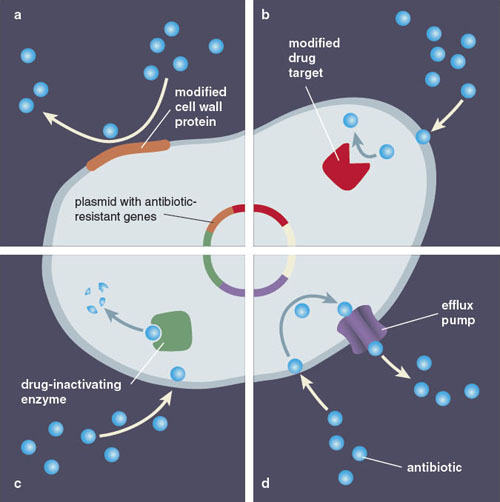
Illustration by Barbara Aulicino
Bacteria in the impermeable-barrier group are naturally resistant to certain antibiotics, either because they lack the target of the antibiotic or because their membranes are impermeable to the drug. In the second group, target modification, bacteria acquire mutations in genes that modify the target of the antibiotic, dampening its effectiveness. Each antibiotic is designed to target a well-defined essential bacterial process. For example, fluoroquinolones are a widely used class of antibiotics for the treatment of skin, lung, or urinary tract infections. These antibiotics target DNA, disrupting the proper functioning of proteins involved in unwinding DNA’s helix during replication. Mutations that confer resistance toward fluoroquinolone antibiotics often change the conformation of these proteins, reducing the binding of the drug to its target and thus increasing the concentration necessary to block the process.
In antibiotic modification, a resistance gene can encode an enzyme that helps break down or modify the antibiotic before it can kill the bacteria. This tactic is often used against beta-lactams, the most widely prescribed and diverse chemical class of antibiotics, which includes the well-known drug penicillin. Penicillin inhibits enzymes that remodel the bacterial cell wall and are essential for the cell during growth. Resistance toward penicillin is frequently conferred by beta-lactamases, enzymes that cleave the penicillin molecule to render it ineffective in inhibiting the cell wall modification enzymes.
Finally, efflux occurs when a resistance gene codes for proteins that actively pump the antibiotic out of the cell, keeping its internal concentration low enough to prevent inhibition. This resistance mechanism is deployed for all antibiotics that have targets within the cell; in many cases such efflux pumps are able to push out several different antibiotics, resulting in multidrug resistance. An example of this has been observed for tetracycline, an agent used to treat a wide variety of infections. Resistance to the drug can stem from tetracycline efflux genes, which code for proteins that sit in the bacterial membrane and export the antibiotic out of the cell.
Further complicating matters, resistance toward any one drug typically results from more than one mechanism. For instance, tetracycline resistance has been observed to occur through target modification, antibiotic modification, and efflux mechanisms.
Though the term antibiotic resistome didn’t emerge until five years ago, the concept encapsulates decades of research on the transmission and evolution of antibiotic resistance genes between different microbes and different environments. The resistome as it is currently defined is the entire suite of genes, from a particular microbe or microbes, which confers antibiotic resistance. It includes all antibiotic resistance genes in a group of microbes at any scale, from a single organism to all of the microbes in an arbitrary environmental sample. Viewed this way, the resistome from one environment can be evaluated for its capacity to exchange resistance genes with another environment.
Mounting evidence indicates that essentially all microbial environments contain antibiotic resistomes; that myriad natural and human-driven activities influence their exchange within and between environments; and that a complex web of interactions connects various resistomes. Two of the most important environments for microbial resistome evolution and exchange are the soil and the human body, but there are other resistomes as well (see the box below).
Antibiotic resistance is everywhere, even in your backyard. Soil microbes likely represent the evolutionary reservoir of most resistance, and the resistome of the soil is easily the largest and most diverse of any environment. The majority of antibiotics used in the clinic were originally discovered as or derived from natural products of soil-dwelling microbes, primarily of the Streptomyces genus of the Actinomycete phylum.
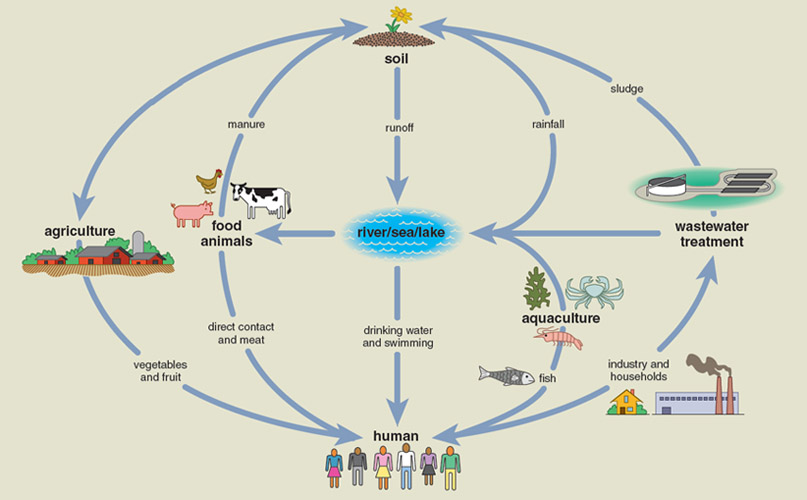
Illustration by Tom Dunne
In 1973, Julian Davies, now at the University of British Columbia, and his colleagues first hinted at the idea of a resistome in the Producer Hypothesis on the origin of clinical resistance. He argued that the “producer” bacteria Actinomycetes, a major source of antimicrobials for the commercial drug industry, must contain elements protecting against the antibiotics they produced. These elements were by definition antibiotic resistance genes. Given that these production capacities likely evolved hundreds of millions of years ago, the corresponding resistance genes are likely just as old.
The antibiotic resistance genes observed in nonproducer organisms (including pathogens) may have been acquired directly from the producers or from their soil-dwelling neighbors who evolved them in response to the selection pressure of natural antibiotic production from other microbes in the soil. In fact, Davies’s group showed the activity of the generally nonpathogenic soil bacteria Streptomyces coded for resistance enzymes that modify specific antibiotics known as aminoglycosides identical to those found in clinical pathogens.
In the 40 years since Davies’s proposal, a large body of literature has provided general support for this hypothesis, as well as some key refinements. A landmark paper from Gerry Wright and his colleagues at McMaster University in 2006, which also formally introduced the resistome, showed that about 400 randomly isolated soil Streptomyces samples were highly multidrug resistant against a large panel of clinically relevant antibiotics. On average, these bacteria were resistant to seven to eight drugs, and one superbug was resistant to 15 different compounds of the 21 tested, including drugs that were entirely synthetic and ones that were only recently approved for clinical use. Wright’s observation was startling, because such high levels of multidrug resistance exceed those found in most pathogens.
In 2008, our own research further expanded the view of a substantial multidrug soil resistome by describing about 600 soil bacteria—from three of the 60 phyla, or divisions, of the bacterial world, namely, the Proteobacteria, Bacteroidetes, and Actinomycetes— that can actually feed and grow on antibiotics. These bacteria were on average resistant to 17 of 18 clinically relevant antibiotics profiled, likely displaying this incredibly high multidrug resistance because they were cultured under the selective pressure of extremely high antibiotic concentrations. Other censuses of cultured soil bacteria populations have since confirmed these high levels of multidrug resistance.
Just one gram of soil is estimated to contain about one billion bacterial cells, and no current method gets even remotely close to sampling this diversity.
The discovery of ubiquitous multi-drug resistance in soil microbes suggests that the soil resistome is immense. Complementary investigations of the genes conferring this multidrug resistance corroborate this prediction. Numerous PCR-based assays (using the polymerase chain reaction to amplify DNA samples) have shown that although resistance genes are already present in virtually any soil, these natural resistomes are likely being enriched by human activity. David Graham and colleagues at Newcastle University analyzed a panel of Dutch soils archived between 1940 and 2008 for both the presence and the abundance of a series of specific resistance gene types. They found a dramatic increase in levels of key beta-lactam, macrolide, and tetracycline resistance genes over the 70-year study period, closely matching the era of large-scale human antibiotic production.
The surveys of known resistance genes in the soil resistome are just the tip of the iceberg. Jo Handelsman, now at Yale University, and her colleagues pioneered the application of culture-independent functional metagenomics, a technique that provides the functional gene composition of environmental samples, to characterize soil resistomes from both human-affected and pristine environments. Using this approach, they identified a number of novel antibiotic resistance genes, some with never-before-seen mechanisms of resistance. Taken together, these studies indicated that the soil resistome was diverse, ancient, and recently enriched by human activity. Direct support for the notion that the soil resistome long predated clinical use of antibiotics comes from recent work from Gerry Wright’s group. In 2011, they reported on DNA sequencing of 30,000-year-old Beringian permafrost that uncovered evolutionary relatives of resistance genes against important modern antibiotics, including beta-lactams, tetracyclines, and vancomycin.
One might expect that this wealth of information on the breadth and depth of the soil resistome would confirm the key prediction of the Producer Hypothesis, that the same resistance genes identified in clinical pathogens would also be identified in soil bacteria, suggesting recent exchange between the clinical and soil resistomes. Surprisingly, such evidence was lacking until very recently. The overwhelming majority of soil resistome studies revealed only limited similarity to resistance genes found in pathogens.
To account for this unexpected result, we hypothesized that a key subset of soil bacteria—the notoriously multidrug-resistant soil Proteobacteria— may represent a conduit for recent exchange with pathogens. We reasoned that since the greatest current clinical burden for multidrug resistance comes from multidrug resistant proteobacterial pathogens, their closest cousins in the soil might show evidence for recent resistome exchanges.
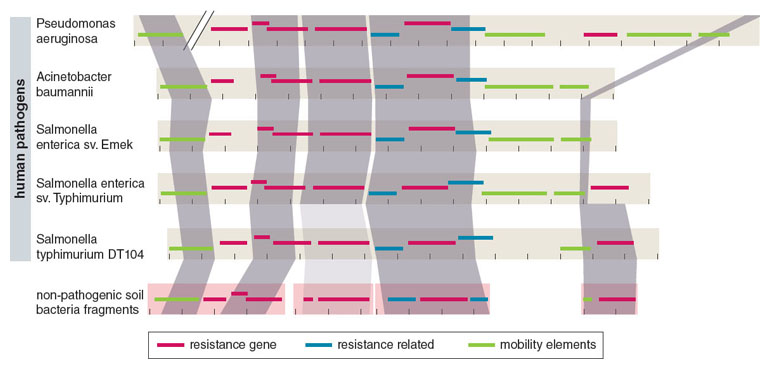
Adapted by Barbara Aulicino from K. J. Forsberg et al., Science 337:1107.
We then set out to test this idea using culture-based selections to selectively enrich about 100 highly multidrug-resistant soil bacterial cultures, composed primarily of Proteobacteria. We profiled their resistomes using a novel approach that integrates culture-independent functional metagenomic selections, next-generation DNA sequencing, and optimized computational sequence assembly and annotation algorithms (see Box 1 above). Through this method, named PARFuMS (for Parallel Annotation and Reassembly of Functional Metagenomic Selections), we uncovered nine different antibiotic resistance genes from diverse U.S. soils that were genetically identical to a number of globally distributed clinical pathogens, finally providing broad support for recent exchange between the resistomes of nonpathogenic soil bacteria and human pathogens.
Despite recent key advances in our knowledge of soil resistomes, we are still in the infancy of exploring this incredibly diverse ecosystem. Just 1 gram of soil is estimated to contain about 1 billion bacterial cells, and no current method gets even remotely close to sampling this diversity. Approximately half of the 60 predicted phyla of the bacterial world cannot be cultured in a lab, and even the ones that can are still not completely characterized. Fortunately, advances in both culture-based and culture-independent experimental methods, as well as improved computational tools for their analysis, enable scientists to scan and sequence the DNA of individual microbes without having to culture them first so they can fill in these holes.
Although the soil resistome is the most important reservoir of resistance from an evolutionary perspective, the microbes living in and on us—known as the human commensal microbiota—incorporate the resistome that is most accessible to human pathogens. These microbes outnumber human cells by 10-fold, and their collective genes (the microbiome) outnumber the genes in the human genome by over 100-fold. Specialized and relatively stable ecosystems of microbes inhabit various parts of the body, with the densest and most diverse community housed in the human intestine. Nearly every aspect of the human condition, in health and disease, involves some interaction with the microbiota and microbiome.
Because one of the microbiota’s main jobs is to keep pathogens from invading the gut, and the fact that any invading pathogen is more likely to interact with bacterial cells than human cells, the commensal resistome is often in a position to exchange resistance genes with nearby pathogens. The increasing levels of antibiotic exposure that the microbiota has been subjected to since the beginning of the antibiotic era provides ample selection pressure to maintain a robust and diverse resistome to participate in these exchanges.
The earliest insights into the human commensal resistome come from culture-based studies of these bacteria. Studies in the 1990s by the University of Illinois’s Abigail Salyers on the normally mutualistic gut microbe Bacteroides established that tetracycline and macrolide resistance genes were being passed back and forth between the resistomes of pathogenic and nonpathogenic forms of the bacteria. Their analysis of archived Bacteroides samples also established that the levels of these resistance genes had steadily increased over the course of two decades.
The implication from these studies—that increased antibiotic use was leading to higher levels of resistance—was supported by numerous other studies on other commensal microbes. Martin Blaser and colleagues at New York University found increases in macrolide resistance in commensal Enterococci, a bug associated with urinary tract infections and meningitis, as a collateral response to therapy targeting another bacterium, Helicobacter pylori, found in some ulcers. They also observed that this enriched resistance persisted for years after therapy ceased, challenging the conventional wisdom that antibiotic resistance encoded a fitness cost to the host bacterium and hence would evolve away or be outcompeted by antibiotic-susceptible strains in the absence of the antibiotic.
These results are not unique to the gut ecosystem; for example, persistently antibiotic resistant Staphylococcus epidermis, a major player in hospital-acquired infections, have been isolated from the nostrils of antibiotic-treated patients. Numerous studies in animal models and in humans show the commensal resistome can readily participate in exchanges within and between ecosystems. Anette Hammerum and colleagues at Statens Serum Institut in Denmark recently demonstrated the transfer of vancomycin resistance genes between human and swine hosts.
All of this evidence indicates that antibiotic treatment selects for genes conferring antibiotic resistance, that these increases in resistance can persist for many years, and that these resistance genes can be exchanged within the commensal microbiota as well as with foreign microbes. As with the soil, we and others in our field have also begun to appreciate that this portrait of the human commensal resistome is a vast underestimate due to an over-reliance on culture-based methods.
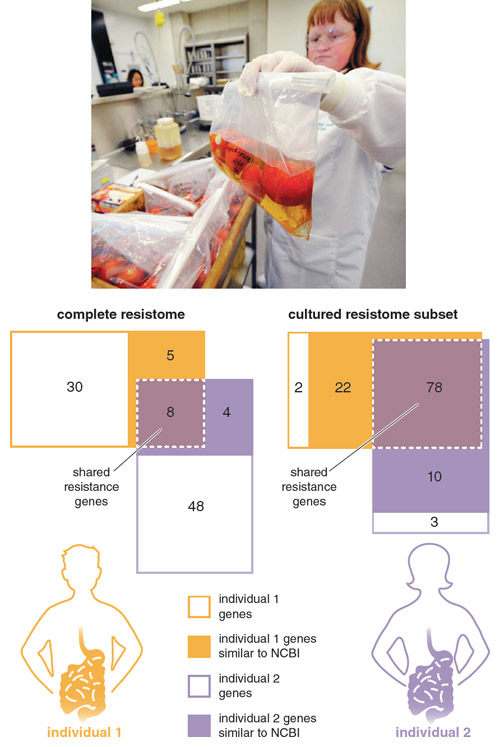
Adapted by Barbara Aulicino from M. O. A. Sommer et al., Virulence 2010 1:299
In 2009 we reported on the first application of culture-independent functional metagenomic selections to study the commensal gut resistomes of two healthy, unrelated individuals, who had been antibiotic therapy–free for at least one year. We characterized the resistomes of both a subset of bacteria cultured from the subjects’ fecal samples (the cultured resistome) as well as that of the entire uncultured bacteria (the complete resistome) from the same samples. We found that the cultured resistome largely consisted of genes found in public sequence databases, and that most of these genes were identical to resistance genes found in common pathogens.
This result confirmed that the resistome of cultured bacteria from the human gut have been in recent exchange with pathogenic microbes. In stark contrast, the majority of genes we identified in the complete resistomes from these same fecal samples were novel, meaning they had little similarity to any genes in public sequence databases. Note that every one of these novel genes enabled the model pathogen E. coli to resist clinically relevant antibiotics, highlighting that this uncharacterized genetic reservoir is fully functional and must be considered when evaluating resistome exchanges.
A couple of recent microbial censuses indicate we have just scratched the surface; deeper interrogation of the resistomes of many more individuals is required to begin to approach a comprehensive view of the human commensal resistome. Earlier this year, research groups led by Peer Bork at the European Molecular Biology Laboratory in Heidelberg and Baoli Zhu at the Institute of Microbiology in Beijing reported on computationally predicted resistomes from sequencing data of human gut microbiomes from 207 and 162 individuals, respectively, representing multiple nationalities and cultural traditions. Collectively, these studies predict the existence of thousands of resistance genes across the analyzed commensal microbiome. The abundance of resistance genes correlates with the available data on the amount of human and agricultural antibiotic use, as well as with how long ago those antibiotics were introduced.
Although we are beginning to gain a glimpse into the genetics of such complex ecosystems, we will need improved culture-based and culture-independent techniques to better understand these reservoirs of antibiotic resistance. Long-term studies of various healthy and perturbed groups of human microbiota will enable us to transition from snapshots to movies of commensal resistomes. Where possible, matched samples are being collected from microbial habitats touched by human activity to map out the ecology and transmission dynamics of resistome exchange. Community-wide microbiome surveys, such as the Earth Microbiome Project and the Hospital Microbiome Project, are profiling the genetic sequences of specialized environmental microbiomes and providing a framework for mapping resistome interactions. Methods such as PARFuMS will enhance the ability of functional metagenomics to uncover novel resistance mechanisms.
The most resounding message that comes through from every new resistome study is that the pool of resistance genes, and the mechanisms of resisting antibiotics, available to bacteria are effectively limitless.
The investigation of resistomes in diverse microbial habitats, including the many not discussed here, serves multiple purposes. There is clearly the basic science interest in understanding the principles that govern the ecology, evolution, and dynamics of antibiotic resistance. Such knowledge will also assist the critical goal of slowing down the spread of multidrug resistance in the clinic and extending the therapeutic lives of antibiotics. But no amount of resistome characterization will end the fight against infectious diseases.
What is most desperately needed are new antibiotics, and as many of them as possible. The most resounding message that comes through from every new resistome study is that the pool of resistance genes, and the mechanisms of resisting antibiotics, available to bacteria are effectively limitless. To stay ahead of the game we must take a multipronged approach, looking for new ways to keep pathogens in check, while also searching for even newer antipathogen strategies. Even when therapies appear to be highly effective during their initial deployment, it is only a matter of time before pathogens tap into the enormous resistomes of their many neighbors and once again thwart our very best drugs.
Click "American Scientist" to access home page

American Scientist Comments and Discussion
To discuss our articles or comment on them, please share them and tag American Scientist on social media platforms. Here are links to our profiles on Twitter, Facebook, and LinkedIn.
If we re-share your post, we will moderate comments/discussion following our comments policy.