How "How It Works" Works
By Felice Frankel
Frank O'Connell of the New York Times discusses his drawings for the newspaper's technology features
Frank O'Connell of the New York Times discusses his drawings for the newspaper's technology features

DOI: 10.1511/2005.51.66
Imagine being handed a gizmo and asked to come up with a clean visual description of what it looks like, what it does and how it works, all fitting neatly into a prescribed space. At the New York Times, Frank O'Connell does just that. I asked Frank about how the illustrations for the paper's popular "How It Works" technology feature come together.
F. F. Tell us, how does the process begin? Is it collaborative between you and the editors or writers?
F. O'C. The process is very much a collaborative effort between myself and Henry Fountain, who is the editor of this feature. Henry has a great design sense.
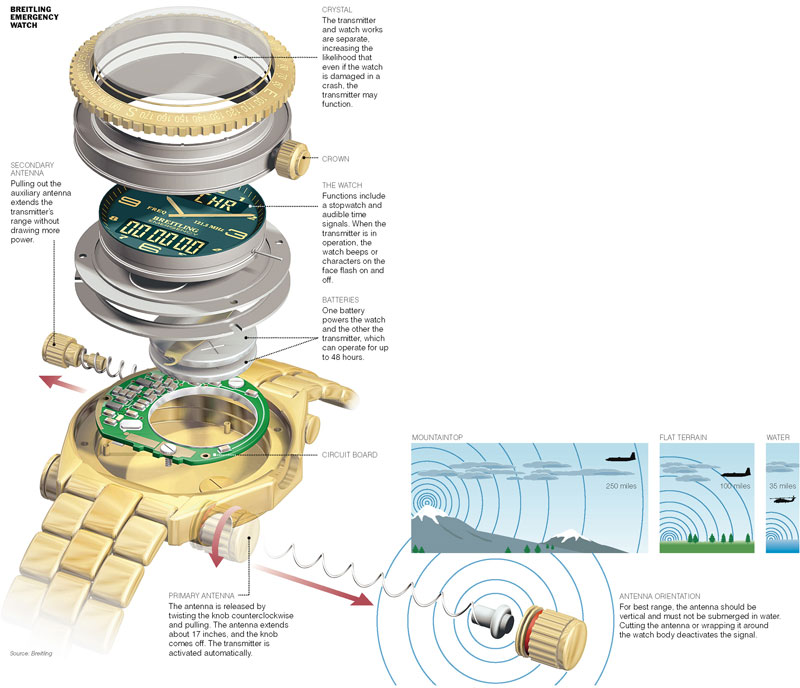
Once we decide on a subject, I'll contact the vendor, explain what we're planning to do and ask the vendor to send us the product. Sometimes this is impractical because of the device's size or value. In that case, I'll request line drawings or digital photographs instead. I always prefer to take apart the object myself, since it gives me a better understanding of how the parts fit together. This way I can assure myself of the accuracy of my illustration. I'll also ask for any technical manuals and media kits the vendor has, so that I can familiarize myself with how the product works.
When the materials arrive, Henry and I meet to discuss which aspects of the product we want to show. In the case of an exploded-view graphic, I often find it preferable to disassemble the device and get the 3-D illustration well under way first. That way I'll know exactly which components I'll have to draw, and I can experiment to find the best positioning and lighting before committing to a layout.
I'll then render the illustration at a low resolution and design my final layout around it, using blocks of dummy text with relevant labels, and go over these with Henry, who writes the captions.
When I first started doing this feature, the story had usually already been written before I began my illustration. I would interact a great deal with the writers and often use their sources. But over the years, the procedure has changed, and more often than not, I do my own reporting, and the graphic is well under way before the story is filed.
F. F. Tell us about your editing process. Do you first create a hierarchy of information?
F. O'C. Henry and I first decide which element will be the main focus. Then we discuss any step-by-step or closeup graphics that will be needed. Because of the amount of space we have to work with, we're able to have a fairly large disparity in size between the main and secondary elements, yet still have the secondary elements large enough to convey useful information.
Not all of our graphics are exploded views or cutaways. Several have involved large outdoor scenes, as when we explained how a bridge de-icing system works. We've also done views of Earth from space, as when we featured the Global Positioning System. But even in these cases, the overall hierarchy of a large dominant visual supplemented by smaller graphics was the same.
Click "American Scientist" to access home page
American Scientist Comments and Discussion
To discuss our articles or comment on them, please share them and tag American Scientist on social media platforms. Here are links to our profiles on Twitter, Facebook, and LinkedIn.
If we re-share your post, we will moderate comments/discussion following our comments policy.