
This Article From Issue
January-February 1998
Volume 86, Number 1
Page 15
DOI: 10.1511/1998.17.15
We are remembered by our exaggerations. So at a recent reunion of my former research-group members, several recalled my saying "When you see a standard deviation in an x-ray crystal structure, multiply it by pi [π, 3.141...], or if the structure is done by friends, by e [2.718...]."
I was talking about structures of molecules—details of their geometry, also of a particularly fruitful way to gain knowledge of these structures. And of the error estimates in such studies.
There is no more basic enterprise in chemistry than the determination of the geometrical structure of a molecule. Such a determination, when it is well done, ends speculation and provides us with the starting point for understanding every physical, chemical and biological property of the molecule. Indeed, the chemical sciences (only modestly imperialistic, I take them to range from materials science through molecular biology) are what they are today largely as the result of careful structure determination. We'd be still waiting in ignorance if we believed the hype of various microscopies. A few very accurate structures have come to us through ingenious use of electron diffraction and various spectroscopies. But the vast majority of what we know about shapes and metric detail of molecules and extended materials derives from studies of the diffraction of x rays by single crystals of molecules, a technique popularly called "x-ray crystallography."
The Intermolecular Shuffle
The molecules in a crystal are not in a vacuum. They are in the solid state because of intermolecular forces—variously called van der Waals, dispersion or crystal-packing forces. Easy to parameterize in approximate calculations, excruciatingly hard to compute a priori, the intermolecular forces are weakly attractive at long distances, but have a repulsive hard core. The tiny attractions add up and make the molecules condense. And, because molecules are to a degree flexible, the intermolecular forces affect distances and angles in every molecule in the crystal by a small step dance of pushing and pulling on each other. Something is gained overall—otherwise the crystal would not form. But crystallization may take place at the cost of a small change in a bond distance or an angle relative to the unencumbered gas-phase structure. There are no tugs on a molecule sailing through outer space.

David Hockney
Ad Right
These weak intermolecular forces are behind my skeptical exclamation. Let me elaborate, and then tell a story of how the very same forces that cause the substantial chemical variance in the structure of a molecule can be used to get valuable information about chemical dynamics, namely the motion of molecules in the course of reaction. Indeed, there is an anniversary to mark—it is nearly 25 years since a pair of landmark papers showed us, with astonishing clarity, that this was possible.
What Crystal Structures Tell
Crystallographic studies are usually done very carefully, although sufficient mistakes of a certain basic type (so-called space-group assignment) are made to provoke one distinguished crystallographer to publish a dreaded article per year replete with his colleague's errors. The crystal structures are carried out by people perhaps more aware of systematic and random error (that "standard deviation") than almost any subculture of our science. So why do I malign their labors?
Actually I don't malign them. I am a voracious consumer of crystal structures—I value them, I treasure them. It's just that the deviations I need and love are not the standard deviations they provide. I want "chemical" estimates of uncertainty: They give me experimental variances.
The standard deviation of a geometrical parameter in a crystallographic structure determination—be it a unit-cell coordinate, a distance, a bond angle—is the square root of a variance, the latter usually denoted σ2 owing to a multitude of experimental uncertainties.
These are accounted for usually quite well by the careful experimentalist, though I wonder today—when the variances are spewed out in an inkling by a computer—whether they are really given as much serious consideration as to origins as they were decades ago.
In my years of molecular voyeurism, I saw the crystal structures get better, and the standard deviations sink, for good data sets to the 0.002 angstrom (Å) [1 Å = 10–8 centimeters] level for organic-molecule distances. But it was also clear to me that ± 0.002Å made no chemical sense. For I saw in some of the structures (technically those with several molecules in the asymmetric unit) molecules that were chemically identical yet whose relevant matching distances or angles differed by much more than the listed standard deviations, because of the intermolecular forces at play. In fact after a while I began to look for these cases. Or to get an estimate of a chemical standard deviation I focused on the part of the crystal structure that was least interesting—that phenyl group in a triphenylphosphine ligand (hundreds of them, and they should be very much alike). And I saw big deviations.
So that's the reason for my flippant π and e factor. What I also record here is a missed opportunity on my part—the information was on hand, and I just quipped. Antonio Martín and A. Guy Orpen of the University of Bristol in the United Kingdom did what needed to be done (and didn't get their idea from me, either). In what is certain to be a classic paper, in 1996 they showed from an examination of thousands of metal-complex structures that crystal-packing forces cause standard deviations of the order of 0.01 to 0.02Å in ligand distances. So, as it turns out, my π was too conservative!
Snapshots Along the Way
The very same crystal-packing forces, which in a given crystal structure make for more chemical uncertainty than the standard deviations lull you into believing, also teach us so much more than what we imagined we could learn from a static structure. For a group of structures, judiciously chosen, can reveal the geometric changes that a flexible molecule undergoes and even the course of atomic motions in reaction.
Michelle Hoffmann and Edward Roberts
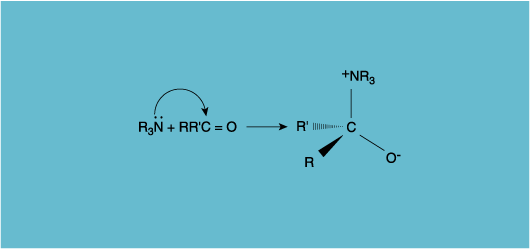
There is a time to praise. I recall so vividly the impact made in this context by two 1973 papers, and I celebrate here their untarnished silver anniversary. In the first Hans-Beat Bürgi, Jack Dunitz and Eli Shefter of the Federal Institute of Technology in Zurich took six structures of reasonably complex organic molecules (perhaps only one of them, notorious methadone, widely known) containing within one and the same molecule an amine (R3N) and a carbonyl group (RR'C=O). These are relatively placid organic functionalities, but they also interact; indeed a ubiquitous organic reaction, a so-called nucleophilic addition of an amine to a carbonyl group, is shown in Figure 2. In solution further steps follow, but the reaction is (and was in 1973) thought to begin as indicated.
Michelle Hoffmann and Edward Roberts
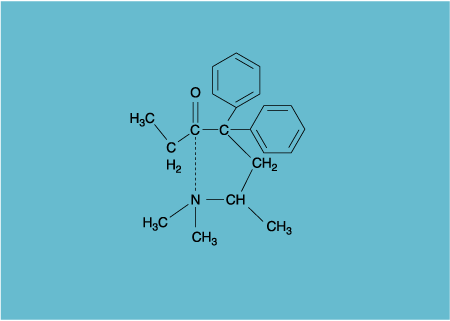
Bürgi, Dunitz and Shefter's six molecules all had the requisite amine and carbonyl in the same molecule. I show only one of the six, the aforementioned methadone (Figure 3). The six molecules are very different, and the packing forces acting on them differ too. Intramolecular flexibility (influenced by strain and conformational restrictions in a given molecule) and variable intermolecular pressures (those packing differences) conspire—no, by beautiful chance, just happen—to "freeze in" the molecules in different points along a reaction pathway. By design, labor and with much luck, the static molecular structures may trace out the way a chemical reaction proceeds, dynamically.
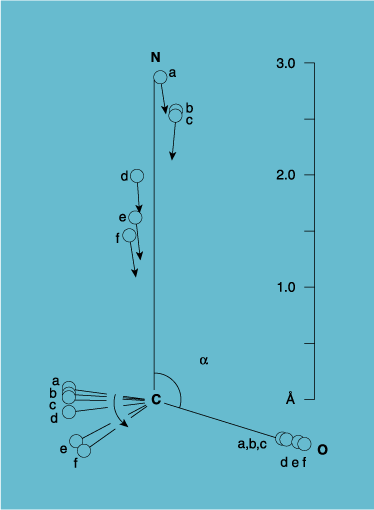
Edward Roberts, from Bürgi, Dunitz and Shefter, 1973.
Many more points have been added to this curve, but I choose to show the six molecules that Bürgi, Dunitz and Shefter plotted on one graph, the fixed carbonyl group pointing to 4 o'clock (Figure 4). See the amine approaching at top? Oh say, can you see the RR' groups on the C of the carbonyl swing down as the amine approaches? We did see, by the x-ray's strong light! The Bürgi, Dunitz and Shefter paper was a true revelation.
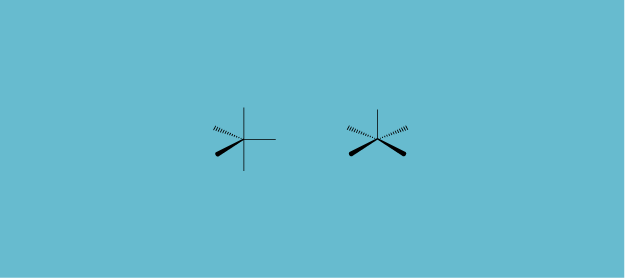
A few months later in 1973, Earl L. Muetterties and Lloyd J. Guggenberger, then at du Pont de Nemours's Central Research Department, looked at a seemingly very different problem. Inorganic molecules in which five ligands are bonded to a central atom can assume a variety of geometries. Most common is a trigonal bipyramid (Figure 5, left). At the time one also had a few square pyramids (Figure 5, right) and a straggling of weird structures not one, not the other.
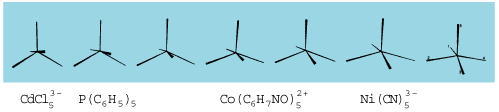
Edward Roberts, from Muetterties and Guggenberger, 1974.
As drawn the two extreme geometries look very different. They're actually pretty close to each other, as R. Stephen Berry at the University of Chicago recognized some time before. A special slight motion of the ligands, named the Berry pseudorotation, takes one into the other. Now look at the crystal structures of seven molecules plotted by Muetterties and Guggenberger (Figure 6).
Cut them out, mount them on cards, flip the cards—you have a movie of the Berry pseudorotation. All that certainty a crystal-clear consequence, as was the beautiful path for nucleophilic addition discovered by Bürgi, Dunitz and Shefter, of the same totality of small forces that make us feel that measured distances might be substantially less certain than indicated.
American Scientist Comments and Discussion
To discuss our articles or comment on them, please share them and tag American Scientist on social media platforms. Here are links to our profiles on Twitter, Facebook, and LinkedIn.
If we re-share your post, we will moderate comments/discussion following our comments policy.