
This Article From Issue
September-October 2012
Volume 100, Number 5
Page 374
DOI: 10.1511/2012.98.374
My first encounter with H2 was typical for a boy in the age of chemistry sets that had some zing to them. My set, made by A. C. Gilbert Co., contained some powdered zinc. It had no acids, but it taught you to generate them from chemicals it included (for instance HCl from NaHSO4 and NH4Cl), or—the manual said—you could buy a small quantity from your local apothecary. Perhaps I got it there, asking politely for the acid in my best accented English a year or so after coming to Brooklyn from Europe. I poured some of the dilute acid on the zinc in a test tube, watched it bubble away, lit (with some fear) a match and heard that distinct pop.
Flammable Air
Next, I encountered the gas, Henry Cavendish’s inflammable air, in a high school electrolysis experiment. We ran a current through water with a little salt dissolved in it, collected the unequal volumes of gases formed, each trapped in an inverted tube. Both gases gave small pyrotechnic pleasures—one, hydrogen, with that satisfying pop when a newly extinguished splint came near it; the other, oxygen, revived exuberantly the flame of the same splint.
Primo Levi, in an early chapter in his marvelous The Periodic Table, describes an initiation into chemistry that features the same experiment, with more fearsome results:
I carefully lifted the cathode jar and holding it with its open end down, lit a match and brought it close. There was an explosion, small but sharp and angry, the jar burst into splinters (luckily, I was holding it level with my chest and not higher) and there remained in my hand, as a sarcastic symbol, the glass ring of the bottom.… It was indeed hydrogen, therefore: the same element that burns in the sun and stars, and from whose condensations the universes are formed in eternal silence.

Image courtesy of the Division of Rare & Manuscript Collections, Cornell University Library.
Ad Right
In my high school lab I had no idea that I was reliving, with different methods, part of the experiment Antoine Laurent Lavoisier thought important enough over two days in February 1785 to invite a select group of luminaries of French science to witness. In a tour de force of the big science of his day, using some remarkable instruments he had constructed at his own expense, he decomposed water into its constituent hydrogen and oxygen, and followed that by a recombination of the elemental gases thus generated into water. Henry Cavendish had proved that water is formed in the combustion of hydrogen some years before; Lavoisier not only decomposed water, but determined that the cycle of its decomposition and reformation proceeded with conservation of mass. Not everyone was convinced—they should have been—yet with this experiment, a new chemical age dawned. Water and air, those seemingly homogeneous elements of the Greeks, were shown to be a compound and a mixture, respectively.
A Diatomic Molecule
Chemistry and I progressed; it took chemistry a good 75 years from Lavoisier’s time to have the macroscopic compounds—there at the beginning, with us today—be joined by a realization of an underlying microscopic reality, imagined well before it was proven, that of molecules. And it took another 65 years (now we’re circa 1925) for the new quantum mechanics to be created, explaining the why and wherefore of the molecules of dihydrogen (a nomenclature I will use when I need to distinguish hydrogen molecules from hydrogen atoms). In my education, I made that transition from compounds to molecules, much as chemistry did. Except I did it in three years instead of 140. I encountered the molecule, more precisely the quantum mechanical treatment of H 2 , in a class George Fraenkel taught, and beautifully so, in my last year at Columbia College.
Fraenkel took us through the first calculation on H2 by Heitler and London, in 1927, a calculation parlayed by Linus Pauling into a general theory of covalent bonding. By this time the dissociation energy of H2 (the strength of the bond, the energy needed to take it apart into two hydrogen atoms) was known. It was 4.48 electron volts (eV) per molecule, 104 kilocalories per mole (kcal/mol). If that doesn’t touch you, let’s begin with the fact that a mole of H2 (roughly 22 liters of it in gaseous form at room temperature) has a mass of 2.0 grams. Not much, that’s why it was used in airships. Kcal/mol? To heat a liter (about a quart, 1.057 quarts to be exact) of water from room temperature to boiling (a real-life operation most of us, even men, have done) takes about 80 kcal. That should help—to knock 2 grams of hydrogen molecules into hydrogen atoms takes about the same energy as to heat one and a quarter liters of water to boiling. Except, don’t try it on your stove—remember the Hindenburg airship.
The energy of the hydrogen molecule as a function of distance is described by a “potential energy curve” shown in the figure below, a graphical depiction of how the chemical potential energy of the molecule varies with separation of the hydrogen atoms (actually their nuclei) in the molecule from each other. The depth of the well relative to the separated atoms is the dissociation energy I described above. But any molecule is a quantum mechanical entity; so the molecule, in a way as a consequence of Heisenberg’s uncertainty principle, does not sit still at the minimum of the potential energy curve. The molecule vibrates, the vibrations of the molecule are quantized—and in its lowest energy state the hydrogen nuclei retain some motion (in a way like a pendulum but less deterministically so) around the “equilibrium distance.” Sometimes they are a little closer, sometimes a little farther apart, on the average they are ~0.74 × 10–8 centimeter, 0.74 Ångström (Å) from each other. We call that the bond distance.
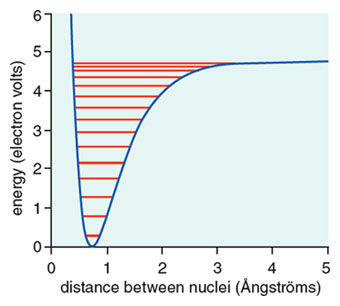
The bond distance in the H2 molecule and its dissociation energy were known by the time the new quantum mechanics came. Heitler and London got a dissociation energy of 3.14 eV, an equilibrium distance of 0.87 Å. Not too great (compared with experiment) but a remarkable result: For the first time quantum mechanics “explained” the existence of a molecule. Which classical mechanics coupled with electrostatics, try as it might, couldn’t.
One could not solve the Schrödinger equation, the wave equation that describes all matter, exactly for H2, but the path down a road of increasingly accurate approximations to the exact solution seemed beautifully logical and enticing to this young apprentice. Fraenkel took us through it first of all by another method, called the molecular orbital (MO) method, pioneered by Friedrich Hund and Robert S. Mulliken. A molecular orbital is a combination of atomic orbitals, an approximate way to describe the location of electrons in a molecule—I will show you one soon. This method eventually dominated chemical thinking from the 1950s through today, but initially gave a poorer description of the H-H bond in H2. Yet both the MO and the Heitler-London methods (expanded into Pauling’s “valence bond” [VB] approach) could be systematically, logically improved. We followed and understood that path in our class, culminating in a remarkable 1933 calculation by H. M. James and A. S. Coolidge, using hand-cranked mechanical calculators, that matched experiment.
I would like to show you the molecular orbitals of H2, because (a) they’re important, and (b) I can’t escape them; they bring to me new chemistry at roughly 25-year intervals. The two 1s orbitals of the individual atoms combine in in-phase and out-of-phase fashion to give molecular orbitals called σg and σu*, shown in the figure below.
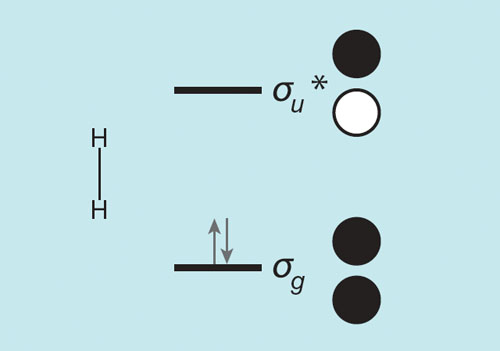
The σ and the subscripts and superscripts on it are labels, symmetry labels; what matters is that σg has no node between the nuclei, while σu* does. That puts σg low in energy, σu* high. And, importantly, σg is a “bonding” orbital, if occupied (as it is in H2), the electrons in it bring the atoms together, whereas σu* is an antibonding orbital, any electrons in it (there are none in an unperturbed H2 molecule, at least in the simpolest analysis) pushing the nuclei apart. Interesting that the big guys, the massive nuclei, move where the small electrons tell them to move.
Treat Me Right
I had a small, almost disastrous encounter with the molecule again, right after earning my Ph.D. Well, spiritually, not materially. An approximate molecular orbital method I and some fellow theoreticians working with W. N. Lipscomb had devised, called the “extended Hückel” theory, did well on some larger, organic molecules, giving reasonable geometries and relative energies. But when I tried it on dihydrogen, the molecule collapsed—the calculated internuclear distance going to zero. That was a shock. It took some courage to go on with a method that could not get right (for good reasons, as we found out) the simplest molecule in the world.
Or, just maybe, this small apparent disaster helped. For it made me and my students rely less on numbers than on understanding. On we did go, and got a lot of chemistry with this deficient method.
A Lousy Acid, a Lousy Base
Molecular hydrogen is pretty unreactive, as is methane. Hydrogen burns, of course (with a flame that is nearly colorless but very, very hot). But to get it to burn you need a match, even though the reaction to form water, Cavendish’s and Lavoisier’s reaction, gives off ~68 kcal/mol of dihydrogen burned. That’s chemistry: Things that should spontaneously proceed by the dictates of thermodynamics (like hydrogen burning) actually encountering substantial barriers to doing so.
Chemical reactivity is predominantly that of acids and bases—that is why we spend so much time in introductory chemistry on this property of molecules. A base (ammonia, for example) is a good donor of electrons; in MO terms it has an energetically high-lying filled molecular orbital. An acid (the hydronium ion, the aquated proton, H3O+) is a good acceptor of electrons, as it has a low-energy empty MO. Hydrogen has an occupied MO, just one; you’ve seen it—it’s the σg in the MO picture of the molecule (see figure above). That MO lies low in energy; H2’s ionization potential, a measure of the energy of that MO, is large, 15.4 eV. And H2’s lowest unoccupied MO, σu*, is relatively high lying—to promote an electron from the filled MO to the unfilled one takes ~11 eV.
Put into plain English, the hydrogen molecule is a lousy base and a lousy acid. The molecule is then relatively unreactive, even as it burns giving off a good bit of heat. Other molecules lack a good handhold, so to speak, on H2.
Döbereiner’s Feuerzeug
Yet hydrogen was known to react all along with some metal surfaces. In another column (American Scientist 86:326–329 [1998]) I recounted how Johann Wolfgang Döbereiner discovered in 1823 that hydrogen burned on a platinum surface. This is the first well-characterized catalytic reaction. Döbereiner did not know that there were molecules in his hydrogen gas (generated the same way I did as a boy, from Zn plus an acid, sulfuric acid in his case). And, of course, he did not know in atomistic detail how those H 2 molecules fell apart on his Pt surface, and how they combined with oxygen from the atmosphere. Döbereiner made a Feuerzeug, a source of fire based on hydrogen, that became a household firelighting tool for half a century.
Molecular Complexes of Dihydrogen
By the 1980s there emerged evidence for weak “complexation” (binding) of hydrogen with various metal atoms. And surface scientists were piecing together the mechanism of Döbereiner’s seeming magic. Elsewhere, organometallic chemists found some reactions in which hydrogen molecules added to a metal center, the two hydrogen atoms split apart in the process. Experimentalists and theorists begin to view the seeming chemical inertness of dihydrogen as a challenge rather than dogma.
In 1984 Jean-Yves Saillard, a French postdoctoral associate (now at the University of Rennes) and I did a careful study of the interactions of hydrogen and methane with discrete transition metal centers with associated ligands. These MLn (M is a metal atom, L a ligand, say CO or PH3, n the variable number of such ligands) fragments, if carefully chosen to be good bases and acids at the same time, could, in our approximate calculations, bind dihydrogen. The molecular orbital essence of our argument is shown in the figure below; a similar picture and interpretation is there in earlier work of three Alains—Dedieu, Strich and Sevin.
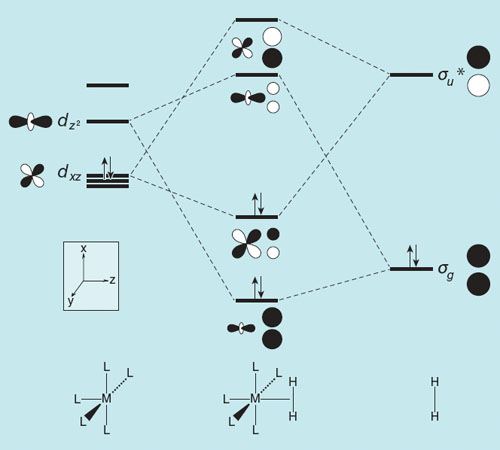
A small interlude here on so-called interaction diagrams, which is what you see in the figure above. These diagrams, my professional bread and butter, show the interaction of the important orbitals of two pieces of a molecule (when it can be taken apart into pieces). That’s the way we build understanding, putting together, in LEGO style, the orbitals of a more complex molecule from simpler pieces. The L5M(H2) molecule in the middle (at that time unknown, at least to us) is built from two simpler pieces—an ML5 fragment at left, and my old friend H2 at right.
The orbitals of H2 are easy—you’ve seen them above, the σgMO, with both of the 1s orbitals of the component H atoms in-phase, at low energy; the σu*MO, unfilled by electrons, at high energy.
On the other side are orbitals of the ML5 fragment, mostly on the metal. They are more complicated (the metal has important 3d orbitals), but the essential feature is that there are orbitals on the metal filled with electrons and some that are empty, and these match in symmetry and overlap reasonably well with the orbitals on the H 2 . The dashed lines in the figure guide us to just these stabilizing interactions.
Here’s what happens in this theoretical analysis: The acid function of the ML5 fragment (its empty orbital, called dz 2) interacts with the base σg of H2, the base function of ML5 (a filled dxz orbital) interacts with the σu*, the acid function of H2. (Did I not say that there is a reason for all that seeming torture on acids and bases in first-year chemistry?) Importantly, there are consequences to the strength and length of the H2 as a function of the interaction: As a result of the mixing of MOs of ML5 with those of H2, some electrons are transferred from the σg orbital of H2, depleting its bonding density. And some electrons are transferred in the opposite direction, from ML5 to the H2σu* orbital. Both actions—decreasing bonding, increasing antibonding—will stretch the H-H distance, even as they overall bind H2 to MLn. The figure is for ML5, but the reasoning extends to other numbers of ligands bound to the metal.
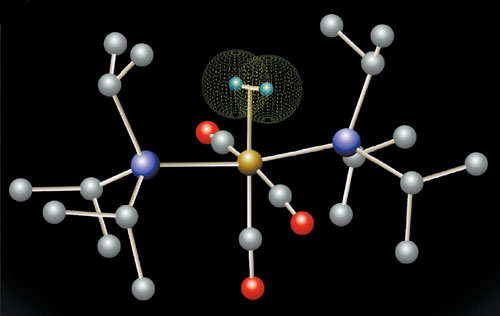
Image by Brian Scott and Josh Smith, courtesy of Los Alamos National Laboratory.
Saillard and I made no prediction of specific molecules. What we did not know when we did our work is that the first such “complex” had just been made. Greg Kubas at Los Alamos had synthesized (and with no nuclear reactions involved), the molecule shown in the figure above. It was followed over the years by a significant group of dihydrogen complexes, even ones in which the metal held more than hydrogen molecule.
In time the H-H distance in these molecules was determined accurately (one needs neutron diffraction for that; metric information also comes from nuclear magnetic resonance studies). Kubas understood very well what was going on—his qualitative thinking about what bound H2 in his molecules, quite independently conceived, was similar to ours.
But what fun for us! A theoretical idea about how a molecule could bind—and not just any molecule, but normally inert hydrogen—translated into reality! We were happy. And Kubas deserves all the credit, because science is ultimately about the reality of a compound in hand—theories come and go, the molecule is there.
The First Element under Pressure
In the past few years, my colleague Neil Ashcroft and I have had a fruitful collaboration on the response of molecules and extended structures to extreme pressure. Three years ago we returned to a first love of Neil’s, hydrogen. In this we were joined by a talented French postdoc, Vanessa Labet.
Experimentally, one can learn much about matter under pressure (see “The Squeeze Is On,” American Scientist 97:108 [2009]) from studies in diamond anvil cells, where in a small reaction volume, between two tough diamonds and enveloped by (one hopes) an unreactive metal, a sample of matter is compressed. At what pressure solid, cold hydrogen (yes, hydrogen freezes, at 14 degrees kelvin) metallizes is the subject of hot, current dispute. But some things people agree on—solid hydrogen retains molecular diatomic units up to pressures such as those at the center of the Earth (3.5 million atmospheres). And from a spectroscopic measurement one can even deduce the internuclear distance in the confined diatomic. As the pressure rises, the H-H equilibrium separation contracts a little, then begins to stretch. The magnitude of the excursion is small, less than 2 percent of the 0.74 Å separation.
There are places in physics and chemistry where theory can afford a clearer picture of a phenomenon, and matter at extreme conditions is one such place. If one can trust the theory.… Vanessa Labet had a numerical laboratory at her disposal of the best structures calculated for compressed H2 by Chris Pickard and Richard Needs. We used that laboratory to get physical insight, to reason out why hydrogen did what it did. The figure below shows the small dance the calculated shortest, intramolecular H-H distance does with pressure—it goes down a little, up for a while, down again, up, down. The discontinuities, the jags in the curve are understandable—they are the consequence of abrupt changes from one preferred form to another, so-called phase transitions. The calculations matched experimental findings pretty well. But what was behind the small dance steps?
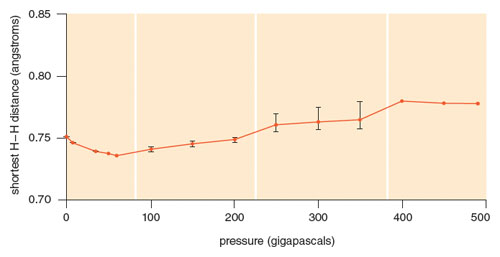
We first thought about the effect of confinement, one hydrogen molecule simply squeezed by other hydrogen molecules in that tense space. Now a model for that was already there in earlier work of Dudley Herschbach and Richard LeSar. They looked at the energy levels of H2 confined in a rigid spheroidal box, as the dimensions of the box decreased. As one might expect, the internuclear separation responded by decreasing. Labet probed confinement by a slightly softer box, a hydrogen molecule imprisoned between two helium atoms, the most ungiving chemical walls we could think of. The earlier results were confirmed—such confinement only made the H2 distance contract. What else could it do?
But that’s not what our numerical laboratory and experiment showed; in a real and modeled crystal of H2 , the hydrogen molecule shrank, expanded, expanded some more, shrank. By just a little. What could possibly make it grow longer ? As it was squeezed ? At this point I remembered Kubas’s wonderful organometallic complexes. In them the coordinated hydrogen molecules expanded, to 0.82–0.89 Å in length. And from the work Saillard and I did, we knew why! The metal fragment provided electrons to populate hydrogen σu*, depopulate σg , both weakening the H-H bond.
In compressed hydrogen, at pressures approaching those at the center of the Earth, there were no metals in sight. But under these extreme conditions, could other hydrogen molecules around a given H2 possibly play that role? We looked at the population of the molecular orbitals of a given molecule, and sure enough the effect was there. Model calculations confirmed that the little dance of H-H separations with pressure that experiment and theory observe in dense, cold H2 was the outcome of two competing effects: simple physical confinement, and the chemical effect of the molecular orbitals of confined and confining molecules interacting, mixing, transferring electrons, stretching that bond. I love it—the same bonding that occurs in discrete transition metal organometallic molecules is there in a highly compressed crystal of pure H2 .
One World
The first element, the simplest diatomic molecule there is—what could be simpler? Hold on—an H2 molecule in solid H2 under pressure, an H2 molecule approaching a Pt surface in Döbereiner’s firelighter, the H2 bubbling out of the solution of a 13-year-old boy playing with slightly dangerous chemicals in a Brooklyn apartment, the H2 in transition metal complexes Greg Kubas saw for the first time in the world—of course, each is different, peculiar, set apart by its conditions of generation and preservation. But there can’t be different rules of nature operating for one H2 and not the other. The joy is in seeing the connections.
Acknowledgment
I am grateful to Neil Ashcroft and Vanessa Labet for making me think about something I never thought I’d be working on again, or that I could imagine there was something left to learn about. As there is.
Bibliography
- Kubas, G. J., R. R. Ryan, B. I. Swanson, P. J. Vergamini and H. J. Wasserman. 1984. Characterization of the first examples of isolable molecular hydrogen complexes, M(CO) 3 (PR 3 ) 2 (H 2 ) (M = molybdenum or tungsten; R = Cy or isopropyl). Evidence for a side-on bonded dihydrogen ligand. Journal of the American Chemical Society 106:451–452.
- Labet, V., R. Hoffmann and N. W. Ashcroft. 2012. A fresh look at dense hydrogen under pressure: 3. Two competing effects and the resulting intramolecular H-H separation in solid hydrogen under pressure. Journal of Chemical Physics 136:074503.
- Levi, P. 1984. The Periodic Table, trans. R. Rosenthal. Schocken Books: New York.
-
- Saillard, J.-Y., and R. Hoffmann. 1984. C-H and H-H activation in transition metal complexes and on surfaces. Journal of the American Chemical Society 106:2006–2026.
- Salem, L. 1987. Marvels of the Molecule (Molécule, la merveilleuse. English), trans. James D. Wuest. VCH: New York.

American Scientist Comments and Discussion
To discuss our articles or comment on them, please share them and tag American Scientist on social media platforms. Here are links to our profiles on Twitter, Facebook, and LinkedIn.
If we re-share your post, we will moderate comments/discussion following our comments policy.