Gene Therapy
By Eric Kmiec
Investigators have been searching for ways to add corrective genes to cells harboring defective genes. A better strategy might be to correct the defects
Investigators have been searching for ways to add corrective genes to cells harboring defective genes. A better strategy might be to correct the defects

DOI: 10.1511/1999.24.240
In the middle of the 19th century, the now-famous monk Gregor Mendel performed his landmark experiments indicating that certain traits can be inherited, and he postulated a discrete unit of inheritance that we now call a gene.
Since then, scientists have come to appreciate how much of an individual's constitution is determined by genes, and, in particular, they have focused on the link between genes and disease. Indeed, over the past 10 or so years, identifying disease-related genes has become something of a cottage industry within the scientific community.
Any reader of newspapers knows just how fruitful this enterprise has been. Almost daily come reports about the discovery of a new gene that contributes to some disease or another, be it sickle cell disease, muscular dystrophy, familial hypercholesterolemia, Alzheimer's or some form of cancer. Right now, therapies directed toward these conditions can only alleviate the symptoms—the manifestations of the defective genes. Implicit, and sometimes explicit, in stories about genetic discoveries is the idea that new therapies can be created that directly address the source of the problem. These gene therapies seek treatments, even cures, that act at the level of the gene itself.
Most of the gene therapy techniques developed so far are of the gene-addition variety; that is, they attempt to provide a good copy of a gene to a cell that harbors a bad one. The hope is that the good, corrective gene will compensate for the bad one and restore the cell to its proper function. Gene addition has been achieved by a variety of means—not only in test-tube experiments, but in clinical trials involving real patients as well. Yet, to date, the results of these trials have been disappointing. Even the most successful clinical trial has fallen short of therapeutic efficacy.
Rube Goldberg, Inc.
Unfortunately, many of these trials have been widely publicized—and, in some cases, oversold—in popular books and magazine articles. Having failed to live up to the inflated expectations created by such publicity, these disappointing trial results have left a general impression that gene therapy cannot now or ever fulfill its initial promise. But these clinical trials may have been conducted before the technology was fully mature, driven in part by investor demands on biotechnology companies to rush products to market. Such clinical trials were almost certainly destined to fall short of the mark.
Many of the fundamental problems with gene therapies have not yet been worked out sufficiently to make the technology therapeutically viable. A number of vehicles have been developed to deliver corrective genes into cells. Some are more effective than others, but none is yet exactly right. The greater challenge, however, lies in the problem of how to make the therapeutic gene behave reliably and at clinically beneficial levels.
Making gene therapy a successful endeavor will require careful research to understand why traditional approaches have not produced the hoped-for results and, in turn, to improve them, while exploring new ways to deal with genetic defects. In my own laboratory, we are considering the possibility that inserting an entire gene into a cell and then expecting it to behave as a native gene may be overly ambitious at this point. It may also be unnecessary. Since the defects in many disease-related genes are fairly small, my colleagues and I are exploring ways to repair rather than replace them. Our initial results lead us to be cautiously optimistic that with adequate basic research this or some other approach will ultimately yield fruit. My own feeling is that pronouncements of gene therapy's imminent demise are as premature as were those overly optimistic pronouncements of its imminent success. At its core, the notion of gene therapy or gene correction is scientifically sound.
Abnormal cell behavior is often the result of an altered gene whose expression is either absent or unregulated. A mutation in just one gene can sometimes cause a cell to malfunction. The mutated gene directs the synthesis of a dysfunctional protein, with the consequence that the cell functions marginally or not at all.
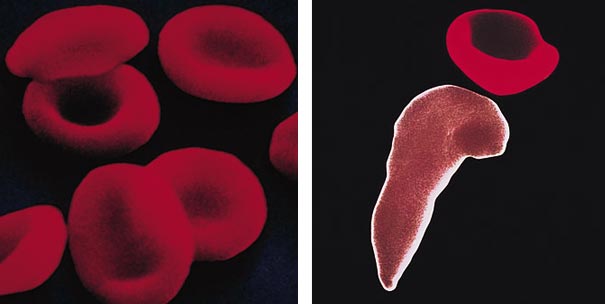
In the case of sickle cell anemia, for example, a mutated hemoglobin molecule actually distorts the red blood cell in which it resides, causing the cell to assume a sickle shape instead of its usual disk shape. The shape change prohibits the cell from adequately performing its designated role of carrying oxygen to the body's organs and tissues.
Another example is presented by muscular dystrophy, which is linked to mutations in the dystrophin gene. This gene codes for the dystrophin protein, which is crucial for the strength and movement of normal muscle tissue. People lacking dystrophin experience the muscle weakness characteristic of the disease.
Finally some genetic mutations do not alter a cell's function as much as they interfere with the cell's normal life cycle, specifically its cell-division cycle. Such mutations can lead the cell to divide uncontrollably, as is the case in certain cancers.
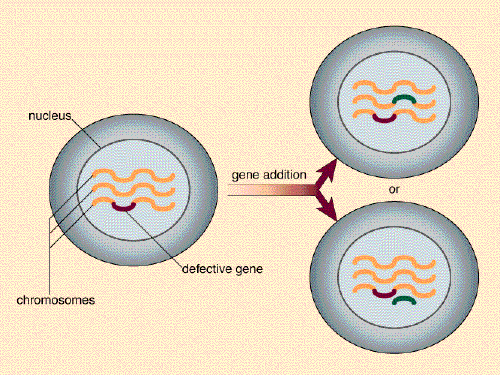
The essence of gene therapy, then, is to deliver to a cell a correct version of a mutated gene, the expression of which will produce the normal protein and hence restore normal cellular function. This has been obvious for some time, but how to achieve this goal has not been. An initial problem centered on how to get a gene into a cell. The chromosomes of a mammalian cell are housed inside a membrane-bounded compartment, called a nucleus. It is not enough for a gene-delivery system to deposit the gene into the cell; the gene must be delivered to the nucleus.
This in itself is not difficult. Scientists have been able to do it for decades. Foreign DNA can be injected into a cell, or its entry can be facilitated by various chemical or electronic means. But these methods are not very efficient, and one requirement for gene therapy is that sufficient amounts of corrective DNA be delivered to enough cells to be therapeutically beneficial.
Under the best circumstances, one would also want the therapeutic DNA to become a permanent part of the host's chromosomes. This would ensure its stability and would mean that the therapeutic gene would be replicated along with the host's chromosomes during each cell division. In contrast, DNA delivered to a cell by physical or chemical means can be placed in the cell's nucleus and can be expressed, but it does not become integrated into the chromosomes.
Edward Roberts
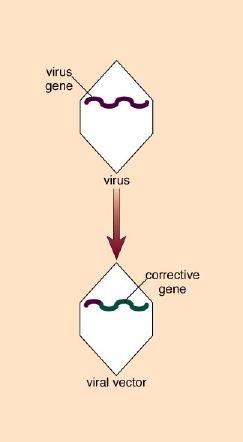
To create these gene-delivery vectors, Mulligan and his coworkers essentially gutted the virus of its genes, disposing of those that could be harmful to the host. At the same time, they retained those genes that enable the retrovirus to insert DNA into host chromosomes. By attaching this integrative machinery to the therapeutic gene, they created a retrovirus capable of infecting cells and splicing a corrective gene into chromosomes.
Edward Roberts
Inserting a gene, however, is only half of the problem. The vector must also contain a mechanism for activating the therapeutic gene, since this is not automatic. Genes have evolved a pattern of expression wherein certain levels of their product are required at specific times in the life cycle of the cell. Hence the corrective action of gene therapy must include a timing and regulatory "device." Such devices are usually found at the start of a gene and constitute the gene's "on" switch, or promoter. But this leads to another problem.

Edward Roberts
Promoters are often exquisitely complex and sometimes quite large, so placing them into a therapeutic vector is difficult. When constructing their retroviral vectors, Mulligan and his colleagues opted to use promoters native to the virus, rather than the corrective gene's own promoter. In laboratory petri dishes, these vectors sometimes worked quite well, but not always.
In some cases, the therapeutic genes entered the cells as expected but were expressed at unpredictably low levels. Low levels of expression continue to dog gene-therapy efforts, and improving expression levels remains a major focus of research. Recent vectors include portions of the gene's own promoter. This has the added benefit that the therapeutic gene is expressed as naturally as possible—only during the times when its product is needed.
Other constructions attach promoters that can be externally controlled. For example, certain genes have promoters that are sensitive to the antibiotic tetracycline and are activated when the drug is present. A vector was recently constructed by Herman Bujold and colleagues at the University of Heidelberg that pairs a tetracycline-sensitive promoter with a corrective test gene. The test gene would be activated only if the patient ingests tetracycline.
The initial expectation was that cells would have to be removed from the body in order to be treated. This ex vivo approach would necessarily limit therapy to those cells, such as blood cells, that are easily removed and replaced. But more recently, retroviral vectors have been developed that can be infused directly into an organ, such as the liver, or placed into the lung by inhalation. This versatility is one of the great advantages of retroviral vectors.
There are also some considerable disadvantages to retroviral vectors that have made investigators cautious about using them. The same feature that makes the retroviruses so attractive to gene-therapy investigators has also been one of their greatest drawbacks—namely the ability to integrate genes into chromosomes. The problem is that scientists have no control over how many copies of the gene become integrated or where on the chromosome they insert. Since integration appears to be essentially random, the vector's genetic payload may become inserted within another important gene, disrupting or altering its expression. Or a gene may integrate within the regulatory region of a gene responsible for controlling cellular proliferation, thus putting the cell on the path towards cancerous growth. Although these are remote possibilities, they are real and must nevertheless be considered as a potential consequence of retroviral-based gene-delivery vectors.
Considering some of the safety issues surrounding the use of retroviral vectors, investigators have been casting about for other viruses that can deliver genes to cells without disrupting their normal chromosomal configuration. There has been much interest in the use of adenoviruses for this purpose. The bulk of the early work on adenoviral gene therapy was conducted by Ronald Crystal at Cornell Medical School and James Wilson at the University of Pennsylvania.
Like the retroviruses, adenoviruses deliver their genetic payload to the nucleus, but, except under rare circumstances, the genes do not integrate into the resident chromosomes. This, of course, relieves concern about random genetic integration, but it also means that the therapeutic gene is only transiently active. The adenoviral vectors have to be repeatedly administered in order to maintain a steady therapeutic dose.
Adenoviruses can infect a broad range of human cells, including those of the lung, liver, blood vessels and brain. In fact, brain tumors have been treated with adenoviral vectors carrying "suicide genes," whose expression leads to cell death only when its product interacts with a specific drug taken by the patient. These studies generated mixed results.
Adenoviral vectors have also been used in human trials to correct mutations in the cystic-fibrosis transmembrane receptor (CFTR) gene, which contributes to cystic fibrosis. The success of these trials, however, has been quite low. For one thing, the host's immune system registers the adenoviral vector as foreign and eliminates it from the system. In addition, some of the vectors cause an inflammatory response at the high levels required to achieve therapeutic doses.
Edward Roberts
One of the most promising vehicles to emerge from recent gene-therapy studies is adeno-associated virus (AAV). This virus infects a wide range of cells, including lung and muscle cells, and it integrates its genes within the host's. In addition, it can infect nondividing cells and does not elicit an immune response—both of which are important advantages over retroviral and adenoviral vectors. The work on this virus has been pioneered by three investigators: Kenneth Berns and Nicholas Muzyczka at the University of Florida and R. Jude Samulski at the University of North Carolina at Chapel Hill. Significant advances in the use of AAV for gene therapy have recently been reported by Mark Kay and colleagues at Stanford University and Kathryn High and coworkers at the University of Pennsylvania. Both of these research groups used a modified AAV vector to achieve long-term expression and correction in animals of a gene that contributes to hemophilia. This achievement required a detailed appreciation for the basic biology of AAV.
However, as expected, this virus has some drawbacks. First, it can carry only a small genetic payload, which considerably restricts its usefulness. Second, it, too, carries the risk of disrupting functioning genes by randomly inserting itself into the chromosomes. Finally, it is somewhat difficult to manufacture these vectors in sufficiently high quantities. Other viruses under study as potential vector candidates include Herpes simplex, Vaccinia and even the human immunodeficiency virus.
In addition to viral-based vectors, investigators are continuing to explore nonviral delivery systems. One system that holds some promise delivers drugs via liposomes, small vesicles artificially created from lipids that resemble those making up the membranes of mammalian cells. Because they are constructed of virtually identical materials, the liposomes can fuse with cell membranes and empty their contents—which can include drugs or corrective genes—inside the cell. Some of the DNA delivered by liposomes makes its way into the cell's nucleus.
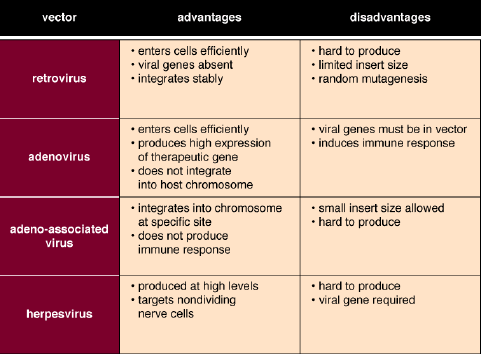
Ultimately, scientists would like to replace a dysfunctional gene with a functional one, within the normal context of the chromosome, an approach that could skirt the concerns about the number of genes delivered, the chromosomal location and the level of expression. Right now, homologous recombination, the only technique that comes close to this, is so inefficient that its success rate is 1 in 10,000. Needless to say, this is not adequate for human use.
Edward Roberts
But the idea of completely replacing a bad gene with a good one may be overreaching, especially when one considers how small are many of the mutations that contribute to disease. To understand how small, we must first consider a few basic facts about the composition of genes.
The gene is to inheritance what a word is to language; it is the basic unit of meaning. In the genetic lexicon, the gene is a length of DNA that codes for a particular protein. The alphabet used by the genetic language contains only five letters, or nucleotides, named for the bases. These are adenine (A), thymine (T), cytosine (C), guanine (G) and uracil (U). The nucleotides A, T, C and G are found in DNA. (RNA, a chemical cousin to DNA and an important participant in genetic decoding, lacks thymine, but has uracil in its place.) The average human gene is a little over 1,000 nucleotides long. In many inherited disorders only one or a few of these nucleotides is incorrect.
For example, sickle cell anemia is the result of a single nucleotide substitution, a single letter misspelled, in the gene encoding the b-globin strand of hemoglobin. Yet this one-nucleotide substitution can cause the structural deformity of the molecule and the characteristically distorted shape of the sickled red blood cell. Over 70 percent of the cases of cystic fibrosis are attributable to the deletion of three nucleotides in the CFTR gene. Why should the entire gene be replaced when the error is so minimal? That strategy seems akin to remodeling the whole kitchen to repair a leaky faucet.
In 1993, while studying homologous recombination in mammalian cells, members of my laboratory began experimenting with ways to repair damaged genes, rather than replacing them. The cell's own repair mechanisms are extremely efficient, as evidenced by the simple and continual inheritance of normal genes through generations of cell divisions. If we could harness the cell's own power of DNA repair, we reasoned, we might be able to correct mutations.
Normal human chromosomes are actually made up of two strands of DNA complexed to each other in an interesting way. It turns out that the nucleotides of DNA can bind with each other in a specific pattern. Except in very rare cases, adenine always pairs with thymine, and guanine always pairs with cytosine. Each DNA strand carries a nucleotide sequence exactly complementary to the other, such that every adenine nucleotide on one strand is matched up with a thymine on the partner strand, and every guanine is matched with a cytosine on the complementary strand. A sequence of GATC on one strand would therefore bind to the sequence of CTAG on its partner, or so it should be.
Occasionally the wrong nucleotide is inserted into a spot, so that the corresponding nucleotide on the partner strand cannot properly bind in that position. In that case the mismatched nucleotides form a bulge. Usually this is not a problem, since the cell contains DNA repair mechanisms that actually scan the DNA and detect such bulges. When one is discovered, the repair systems work to remove the incorrect nucleotide and replace it with the correct one. But if the mismatch is overlooked by the cell's repair machinery, the error is retained, and the gene remains defective.
Edward Roberts
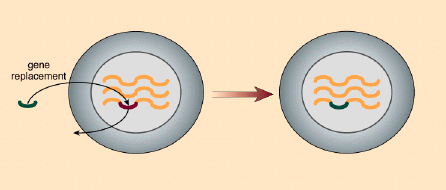
It was our idea to alert these repair mechanisms to the error. The principle is quite simple. We artificially create a short string of nucleotides, called an oligomer, that, with one exception, is exactly complementary to the section of the gene in which the error is located, the exception being at the site of the error. Here we insert the nucleotide complementary to the one that is supposed to be in the DNA sequence of the normal gene. The oligomer binds to its complementary sequence on the DNA, and by design creates a bulge at the site of the mismatch. This bulge is eventually detected by the cell's internal DNA-repair mechanisms. Repair enzymes remove the erroneous nucleotide and replace it with a nucleotide complementary to the one in that position in the oligomer, which happens to be the correct nucleotide. This scenario for targeted gene repair has been experimentally confirmed in my laboratory by Allyson Cole-Strauss.
Our work builds on earlier studies from Fred Sherman's laboratory at the University of Rochester, who used a similar technique to change a single nucleotide. But the oligomers used by the Sherman group were unstable, and the team never extended its work. It turns out that mammalian cells contain enzymes that either degrade the ends of DNA molecules, or link them in long arrays called concatamers, which essentially destroys the integrity of the oligomers. We discovered that we could increase the stability of an oligomer by attaching segments of RNA to each of its ends. Like DNA, RNA is also composed of strings of nucleotides and therefore can bind to DNA in the same complementary manner as can another strand of DNA. (RNA contains no thymine. Instead, the uracil in RNA pairs with the adenine in DNA.)
In the past two years, we have successfully corrected seven chromosomal targets with this approach. Cole-Strauss and others in my lab have demonstrated the feasibility of using gene repair to correct the sickle-cell mutation in vitro, and Clifford Steer's laboratory has reproduced and extended those results in certain animal models. Vitali Alexeev and Kyggeon Yoon have shown that the correction is maintained through successive generations of cell division, suggesting that gene repair may have long-term benefits. Only continued studies will be able to determine whether this approach will be useful for human gene therapy.
In the clinic, gene therapy has enjoyed few successes and many failures. But within these failures lessons have been learned. In some cases, scientific rigor was sacrificed in order to bow to financial pressures to rush gene therapy into clinical trials. In other cases, the goals were too lofty, and the expectations were unrealistically high.
Limited success in animal models all too often leads directly to clinical trials. But a mouse is not a small human with four legs, and the positive results in mice do not necessarily portend a positive outcome in people.
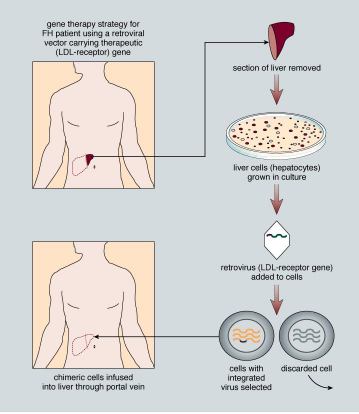
Even at their best, the results of trials with human gene therapy are equivocal. For example, let's consider a gene therapy trial to treat familial hypercholesterolemia (FH). People with this inherited condition have dangerously high blood levels of cholesterol, in spite of their body weight or diet. The condition results from a defective gene that encodes a receptor found on the membranes of liver cells specific for low- density lipoprotein (LDL), what many call "bad cholesterol." Normally LDL enters liver cells via this receptor, after which the liver clears the body of LDL. But people with FH have too few functioning receptor molecules and cannot remove LDL from their blood. As a result, blood serum levels of LDL are too high in people with this condition, and many FH patients develop coronary artery disease.
Photograph courtesy of Eric B. Kmiec
In animal models, investigators demonstrated some success when corrective copies of the receptor gene were transferred into liver cells via a retroviral vector. Blood levels of LDL in the treated animals were significantly reduced and remained so for over six months. These experiments were done carefully, in several animal models and with rigorous controls. All the models produced similar, encouraging results. Based on these results and the fact that patients with this disease have few good treatment alternatives, a human gene-therapy trial for FH was approved.
The experience of one 28-year-old woman represents one of the better outcomes of this clinical trial. The patient lacked any detectable functioning LDL receptor because she lacked the gene for it. At the start of the trial, she had 482 milligrams of LDL in each deciliter (mg/dl) of blood, well over twice the normal level of 160 to 210 mg/dl. Her liver cells were then treated with a retroviral vector containing the LDL-receptor gene. Within a few days, her serum cholesterol dropped by 180 mg/dl to about 300 mg/dl. With additional cholesterol-lowering drugs, her LDL blood levels stabilized at around 356 mg/dl and remained there for about two and a half years. These levels, although lower than they were originally, are still higher than they ought to be.
Herein lies the quandary of human gene therapy: It "sort of" works. This trial demonstrated the feasibility and safety of gene therapy for treating FH. But the results hardly constitute a ringing endorsement for this approach as the definitive therapy. In fact, it would be difficult to name a clinical trial to date that does, a situation that prompted review of the approval process for clinical trials.
Edward Roberts
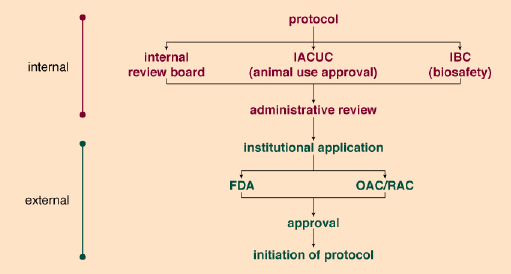
Decisions on clinical applications of gene-therapy approaches fall under the auspices of the federal government. Originally, the Recombinant DNA Advisory Committee (RAC) was charged with the duty of making suggestions for approval or disapproval to the director of the National Institutes of Health (NIH). The RAC was empowered to consider somatic gene-therapy protocols only; that is, the RAC considered protocols that did not involve the so-called germ line cells, such as eggs and sperm. In addition to submitting protocols to the RAC, investigators submitted new gene-therapy protocols to the Food and Drug Administration for Investigational New Drug (IND) approval. More than 100 protocols have been approved by the RAC to date.

Edward Roberts
In 1996, Dr. Harold Varmus, the director of the NIH, amended the procedure because of rising concerns over the rate of failures among approved clinical trial protocols. The fallout of this effort has been an enhanced oversight role for the NIH through three mechanisms. First, the office of Recombinant DNA Activities Advisory Committee (OAC) evaluates protocols. Next, gene-therapy conferences are held to promote public discussion of scientific merit and ethical aspects of proposals for human clinical trials. Finally, the public is informed about the progress of ongoing trials. The gene- therapy policy conferences have already helped to improve the oversight of the approval process. They also encourage continued review of the ethical aspects of each trial. Although in most cases, increasing government involvement is viewed as an invasion into scientific enterprises, gene therapy will stand to benefit by such heightened control.
In spite of the increased government scrutiny of previous gene-therapy protocols, the pressures to bring gene therapy to the clinic do not seem to abate. This is in part because of the formation of ventures aimed at commercializing products and techniques. Financial pressures and constraints often force biotechnology companies to forgo basic research in favor of application-driven development. Unfortunately, this means that promising but technically difficult approaches may never be adequately developed for lack of research funding.
Edward Roberts
I wish to thank current and former members of my laboratory for their hard work on gene repair, the NIH for funding these projects and Michelle Hoffman for editorial advice.
Click "American Scientist" to access home page
American Scientist Comments and Discussion
To discuss our articles or comment on them, please share them and tag American Scientist on social media platforms. Here are links to our profiles on Twitter, Facebook, and LinkedIn.
If we re-share your post, we will moderate comments/discussion following our comments policy.